How Do PROTACs Actually Get Into Cells?
Big molecules, big permeability issues. All medicinal chemists know that well.
No wonder that many scientists in drug discovery were skeptical whether such large molecules (800+ Da) can make it through clinical trials. Yet they enter cells as well as systemic circulation sufficiently to drive potent target degradation and elicit a therapeutic effect. However, how they actually get through the cell membrane has been insufficiently investigated.
Last year brought one piece of the puzzle. The fatty acid transporter CD36 was identified as a receptor that PROTACs need for cellular uptake. But that just moved the question one step deeper: once a PROTAC binds CD36 at the cell surface, how does the whole complex actually get internalized?
A new preprint from Hao-Yang Liu, Jeanne C. Stachowiak, Hong-Yu Li and colleagues (UT Austin and UT Health San Antonio) provides new insights. PROTACs hitch a ride into cells through clathrin-mediated endocytosis (CME), the same general pathway that pulls in many other cargo proteins.
CD36 sits at clathrin-coated pits
The authors used SUM159 cells in which AP2 (the main adaptor of the clathrin pathway) was tagged with a fluorescent label so that endocytic sites could be tracked live. They then expressed CD36 alongside two control proteins: one that is known to enter cells through CME (TfR-YTRF) and one that does not (TfR-CTRD).
Live imaging showed that CD36 forms bright puncta that overlap strongly with AP2-marked endocytic sites. Quantitatively, CD36 recruitment matched the positive control almost exactly, while the negative control stayed well below. In other words, CD36 behaves just like a classical clathrin pathway cargo.
What part of CD36 actually does the work?
To find out, the authors mutated key residues in the cytosolic tail of CD36 and tracked how each change affected recruitment to clathrin sites:
- Ub-mut (K469R/K472R): blocks ubiquitin attachment
- Tyr-mut (Y463A): disrupts a tyrosine motif that AP2 likely reads
- Ub/Tyr-mut: both at once
- C-term truncation: removes the entire tail
Each successive change weakened recruitment. Fitting the data to a binding model gave clear numbers. The Ub-mut increased the dissociation constant 2.6-fold, the Tyr-mut 3.4-fold, and the dual mutant 4.5-fold. The dual mutant also lost 62% of its capacity, and the truncation essentially abolished recruitment.
The takeaway is that CD36 does not have one strong handle for the clathrin machinery. Instead, it relies on several weak interactions working together (probably AP2 reading the tyrosine motif, and Eps15 or Epsin grabbing the ubiquitin tags). This is likely why this pathway was missed before: people were looking for stronger, more obvious motifs.
Tracking a fluorescent PROTAC
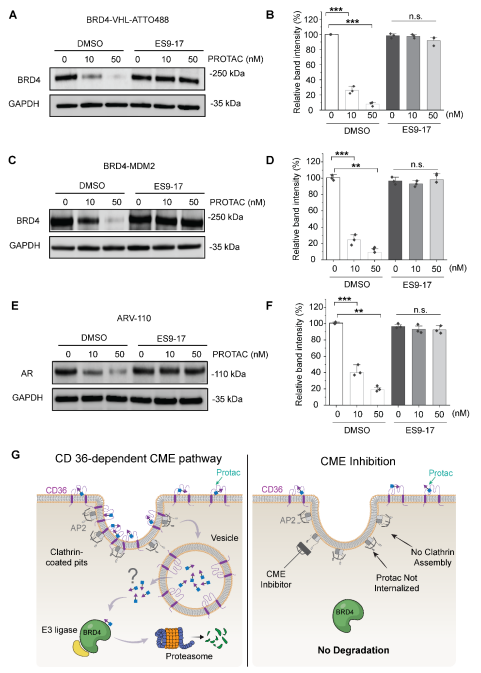
To see what PROTACs actually do at the cell surface, the team made a fluorescent version: BRD4-VHL-ATTO488. This is a JQ1-VHL PROTAC with an ATTO488 dye on a PEG linker. Imaging showed clean colocalization of the PROTAC, CD36, and AP2 at endocytic sites.
When CD36 was knocked down with siRNA (around 90% knockdown by western blot), PROTAC recruitment to clathrin-coated sites dropped by 86%. So CD36 really is the main entry point that delivers PROTACs into the clathrin pathway.
Blocking clathrin blocks PROTAC uptake
The authors then asked the obvious follow-up: what happens if you simply shut down clathrin-mediated endocytosis? They tried two ways. The first was genetic, by expressing AP180ct, a dominant-negative fragment that ties up clathrin in the cytosol. The second used small molecule probe ES9-17 that acts as a clathrin assembly inhibitor.
Both methods gave the same result. By flow cytometry over 2 hours, and by live-cell imaging at 0, 30, and 60 minutes, PROTAC uptake was almost completely lost. Transferrin (a classic CME cargo, used as a positive control) behaved the same way. PROTACs simply do not accumulate inside cells when CME is off.
Without CME, no target degradation
In one of the key experiments the authors tested three different PROTACs covering two targets and three E3 ligases:
- BRD4-VHL-ATTO488 (targets BRD4, recruits VHL, labelled with a dye)
- A1874 (targets BRD4, recruits MDM2)
- ARV-110 (targets Androgen Receptor, recruits CRBN)
In DMSO controls, all three PROTACs worked as expected. But upon pretreatment with 10 µM ES9-17, the degradation was abolished. Genetic CME blockade with AP180ct gave the same outcome. From these results it seems that the dependence on clathrin-mediated endocytosis is not specific to one target or one ligase but rather a general mechanism for smuggling PROTACs into the cells.
My comments
The most interesting aspect, in my view, is the potential generality. Nevertheless it would be interesting to see more PROTACs being evaluated. Of note, one of the protacs (BRD4-VHL-ATTO488) was labelled with dye and the MDM2 PROTAC (A1874) contains large lipophilic MDM2 ligand, meaning that only one PROTAC (ARV-110) represents a “typical drug-like” PROTAC molecule. Therefore, the requirement for clathrin-mediated endocytosis and its generality across targets and ligases would deserve further investigation.
It is well known that PROTAC potency varies between cell types. CME mediated endocytosis of PROTAC can partially explain this observation as CME is cell-line dependent and often upregulated in cancer cells.
If PROTACs need a strongly internalized receptor as a handle, why stick to CD36? As the authors suggest, engineering bifunctional molecules that engage other clathrin cargo (transferrin receptor, RTKs, GPCRs, immune receptors) could in principle deliver them selectively to specific tissues.
A limitation worth keeping in mind is that the paper does not address how PROTACs escape from endocytic vesicles into the cytosol.
Share your opinion
Have you noticed cell-line variability in PROTAC potency that might trace back to differences in clathrin-mediated endocytosis? And do you think receptor hijacking beyond CD36 is realistic for next-generation degraders?
Leave your comment under my LinkedIn post here.
Full preprint: https://doi.org/10.64898/2026.04.06.716751