Biocatalytic Cascades Enable Manufacture of the Macrocyclic Peptide Enlicitid
Enlicitide decanoate (MK-0616) is a beautifully complex macrocyclic (not so small) peptide (or rather pseudo-peptide). This 1720 Da heavy beauty with 10 stereocentres is an inhibitor that blocks interaction between PCSK9 and LDL receptor and is under development at Merck for atherosclerotic cardiovascular disease. Most macrocyclic peptide drugs have to be injected. Enlicitide, however, is orally bioavailable.
The molecule is an octapeptide with six non-natural amino acids, 12 amide bonds, a six-carbon linker, and three fused macrocyclic rings. The original clinical synthesis took 43 steps (!!!), while nearly half of them just protecting group manipulations, and culminated in a ruthenium-catalyzed ring-closing metathesis run at 5 g/L to suppress oligomers, highlighting the complexity of the previous synthetic route.
In a recent Science paper, the team at Merck led by Artis Klapars, Anna Fryszkowska, and Stephanie Galanie came up with a completely different approach. Using seven engineered enzymes and three auxiliary ones, they assemble enlicitide in three cascade steps, with 39% overall yield, no chromatography, and almost no protecting groups.
Retrosynthetic plan
The molecule was broken into three fragments: Western, Eastern, and Northern. The Western and Eastern fragments were tractable by classical chemistry. The Northern fragment is the difficult one, a macrocyclic tetrapeptide with three unnatural amino acids and lots of competing reactive functional groups, where the enzymatic chemo-selective transformations play the central role.
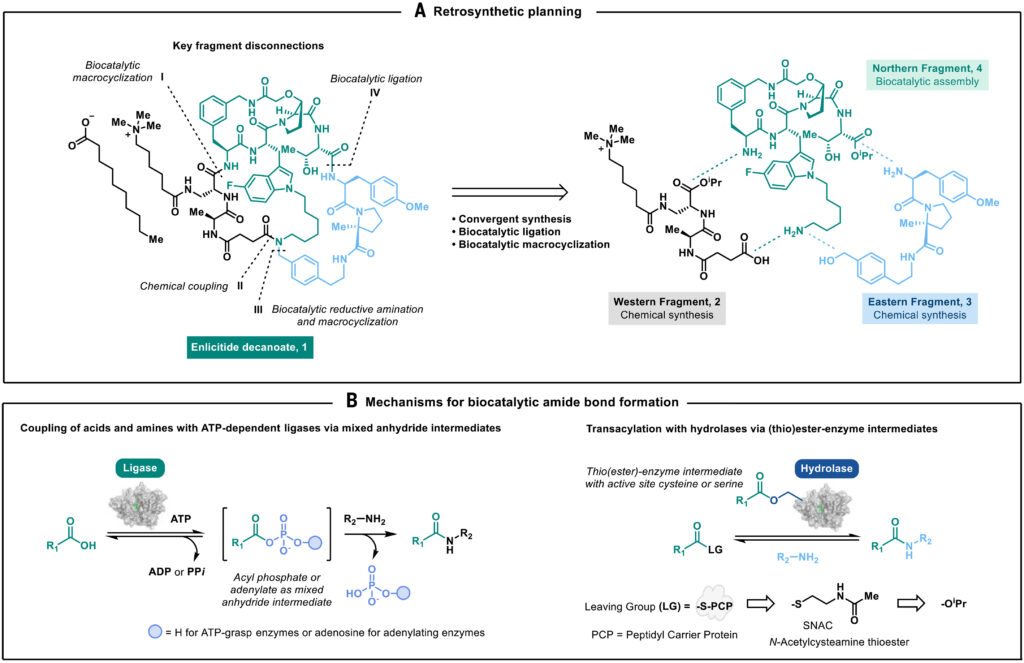
(A) Retrosynthetic analysis of enlicitide 1 prioritizing simplicity, convergence, and key selective transformations catalyzed by enzymes. (B) Two distinct mechanisms of biocatalytic amide bond formation: direct, ATP-dependent ligation of unactivated amino acids through mixed anhydride; ATP-independent transacylation using (thio)esters or amides as electrophiles.
Building the Northern fragment
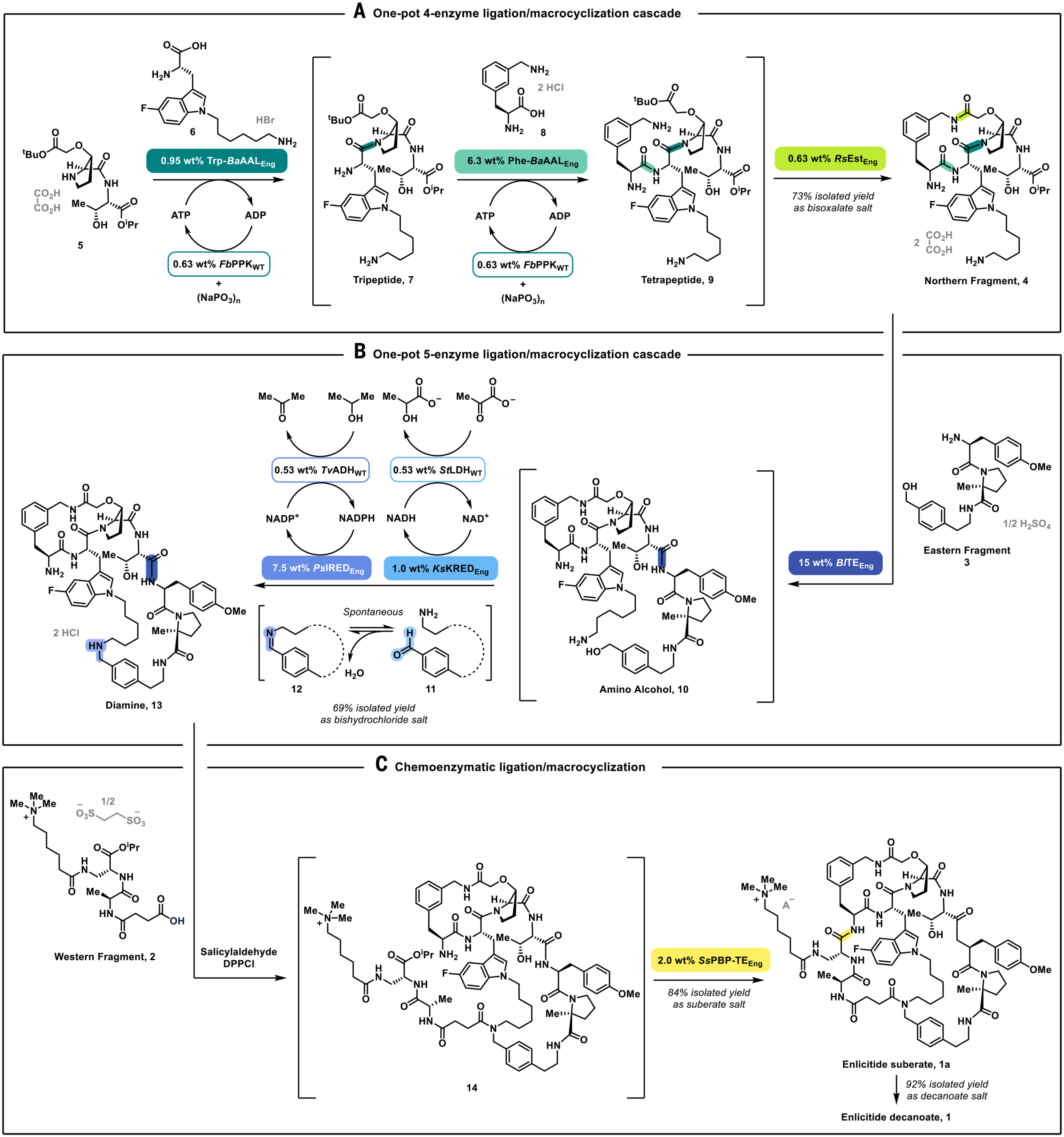
The synthesis starts with a biocatalytic cascade exploiting ATP-grasp ligases from Bifidobacterium adolescentis (BaAAL), that allow direct amide coupling with excellent chemoselectivity, minimizing the use of protecting groups. Of note, the ATP activates the carboxylic acid as a mixed anhydride. The authors divergently evolved BaAAL into two variants: one tuned for the tryptophan-based moiety, one for the phenylalanine-based moiety.
In a single pot, both ligases work side by side. They take a dipeptide and add two more amino acids in the right order, tolerating the other amines and carboxylates floating around. ATP gets recycled by a polyphosphate kinase using cheap hexametaphosphate. As a result, the tetrapeptide has been obtained with an impressive 88% yield.
To the same pot, the authors added an engineered esterase from Roseibacillus sp. (RsEstEng), which closed the first macrocycle at the C-terminal tert-butyl ester which served as a leaving group. After crystallization, the Northern fragment has been isolated with 73% yield on 52 kg scale with no detectable oligomers.
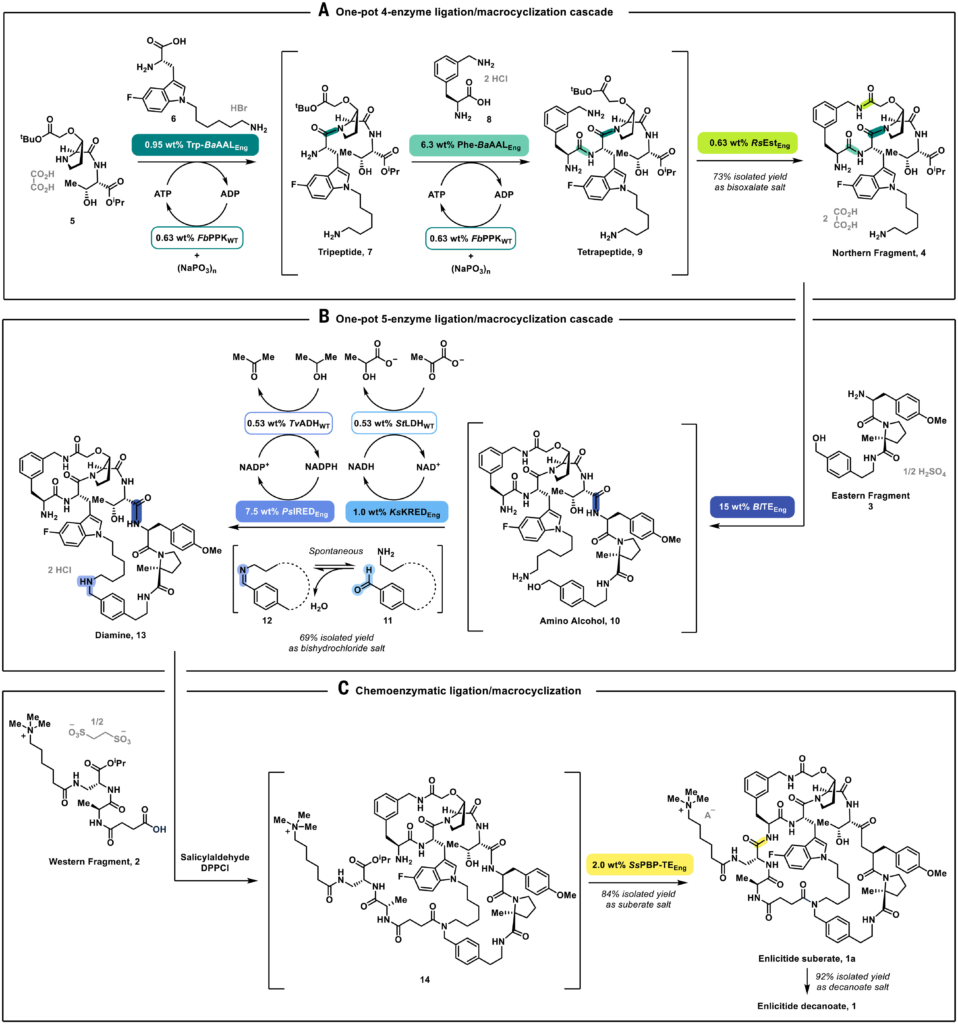
Engineered enzymes (Eng) are in colored boxes and WT auxiliary enzymes are in white boxes. Conditions: (A) Dipeptide 5 (1.1 equiv, ~76 mM), amino acid 6 (1.0 equiv, ~69 mM), amino acid 8 (1.0 equiv, ~69 mM), AMP•H2O (0.10 equiv), Trp-BaAALEng (0.95 wt%), Phe-BaAALEng (6.3 wt%), FbPPKWT (0.63 wt%), MgCl2 (1.33 equiv), sodium hexametaphosphate (6.0 equiv), tergitol 15-S-9 (0.95 L/kg), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer in water adjusted with NaOH to pH 7.5, 25°C, 48 hours, 88 area% of 9 by liquid chromatography (LCAP); then RsEstEng (0.63 wt%), pH 8.0, 25°C, 12 hours; 88% LCAP of 4, 73% isolated yield as bis-oxalate salt on a 52 kg scale. (B) Northern fragment 4 (1.0 equiv, ~ 50 mM), Eastern fragment 3 (1.0 equiv, ~50 mM), BlTEEng (15 wt%), Ca(OAc)2•H2O (1.9 equiv), 2-carboxybenzaldehyde (0.13 equiv), N,N-dimethylacetamide (DMAc), HEPES buffer in water adjusted with NaOH to pH 7.9, 15°C, 32 hours, 89% LCAP of 10; then PsIREDEng (7.5 wt%), KsKREDEng (1.0 wt%), StLDHWT (0.53 wt%), TvADHWT (0.53 wt%), NAD+ (0.02 equiv), NADP+ (0.02 equiv), isopropanol (iPrOH) (3.0 equiv), sodium pyruvate (2.0 equiv) charged over 24 hours, pH 7.8, 32°C, 24 hours; 84% LCAP of 13, 69% isolated yield as bis-hydrochloride salt on a 46 kg scale. (C) Chemical coupling: Diamine 13 (1.0 equiv), salicylaldehyde (1.2 equiv), triethylamine (Et3N) (5.3 equiv), acetonitrile, 0°C, 1 hour, then Western fragment 2 (1.1 equiv), diphenylphosphinic chloride [(DPPCl), 1.35 equiv], 0°C, 1 hour, then liquid-liquid extraction to provide ~44 mM solution of 14 in water-acetonitrile, then enzymatic macrocyclization: SsPBP-TEEng (2.0% wt), 3-(N-morpholino)propanesulfonic acid (MOPS) buffer adjusted with NaOH to pH 7.6, 22°C, 18 hours; 93% LCAP of 1, 84% isolated yield as enlicitide suberate 1a on a 2.5 kg scale, which was converted into enlicitide decanoate 1 in 92% isolated yield.
Combining the fragments together
To combine the Northern and Eastern fragments, the authors employed thioesterases which nature uses in non-ribosomal peptide synthesis to close macrocycles. Specifically, they chose a thioesterase from Brevibacillus laterosporus and swapped the catalytic serine for cysteine to favor amide formation, and tuned it to accept a more stable isopropyl ester instead of the unstable thioester nature uses.
Without isolating the intermediate, the second macrocycle was also closed via enzymatically catalyzed reductive amination. A ketoreductase was used to oxidize a benzylic alcohol to an aldehyde, which spontaneously formed a cyclic imine, and an imine reductase captured it as the cyclic secondary amine. Two wild-type recycling enzymes kept NAD and NADP cofactors turning over using pyruvate and isopropanol as sacrificial reagents. This five-enzyme cascade delivered the bicyclic diamine in 69% isolated yield on 46 kg scale.
To install the Western fragment, authors used salicylaldehyde to temporarily mask the primary amine of the diamine 13, and diphenylphosphinic chloride to activate the carboxylic acid on the Western fragment.
The final macrocyclization step was catalyzed by an engineered penicillin-binding-protein-type thioesterase from Streptomyces sp. (SsPBP-TEEng). Notably, the final ring is closed at 70 g/L, more than 10 times the concentration of the original RCM step (5 g/L), with only 0.1% dimerization and 1.8% ester hydrolysis. After crystallization as the suberate salt (84% yield) and conversion to the decanoate (92% yield), enlicitide was obtained with excellent purity (above 99%) on multikilogram scale!
My comments
Looking at the complexity of the molecule, I can hear many of my colleagues saying that this will never become a successful drug. However, this story is a great testament that modern science is capable of finding remarkable solutions, such as cutting the 43-step-long synthetic sequence down significantly. Once the three building blocks are obtained (each of them can be made in a couple of steps), it takes “only” three enzymatic cascades and salt conversion to finish the synthesis.
This is one of the most impressive applications of biocatalysis to drug manufacturing I have seen in a while. Although I would argue against the authors that the synthesis is not protecting-group free as they are selling it (the tert-butyl ester on the dipeptide, the isopropyl ester on the Northern fragment, and the salicylaldehyde imine masking of the primary amine), the efficiency of how the molecule is assembled is remarkable which is what actually matters the most.
What is less visible from the paper itself is the scale of enzyme engineering investment behind it. Developing seven engineered enzymes is not a task for Friday afternoon, each one likely went through many rounds of directed evolution. Nevertheless, in cases where a compound is produced repeatedly and efficiency matters (such as in drug manufacturing), the invested effort can pay off. We can use it as inspiration for future design (use of ligases or esterases for clean amide coupling, run macrocyclizations enzymatically at high concentration). I expect we will see more such approaches in the future.
Share your thoughts
How do you see biocatalytic cascades fitting into (peptide) drug manufacturing? Will they become increasingly common?
Leave your comment under my LinkedIn post here.
Full paper: https://www.science.org/doi/10.1126/science.aed8713