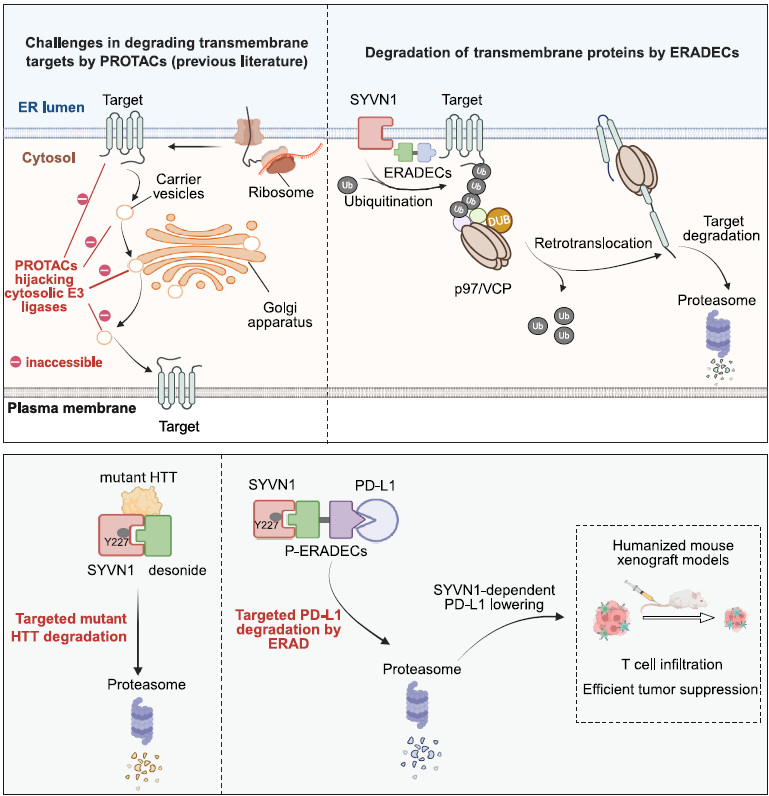
Hijacking ERAD for Targeted Degradation of Transmembrane Proteins
Although PROTACs hijacking cytosolic E3 ligases have been reported to degrade certain trans-membrane (TM) proteins (including EGFR, GPCRs, and PD-L1), It is plausible that this approach is for some TM targets sub-optimal as TM targets have restricted access to the cytosolic degradation machinery. Strategies specifically designed for membrane proteins, such as LYTACs, GlueTACs, and TransTACs, rely on the endosome-lysosome pathway and are therefore influenced by recycling endosomes that tend to shuttle TM targets back to the membrane. Moreover, most of these strategies employ large biomolecules like antibodies with well-known limitations in delivery, cost, and immunogenicity.
Haikun Song, Wei Wang, Tingfang Mei, and colleagues from Fudan University and Naval Medical University (corresponding authors Boxun Lu and Chunquan Sheng) now report in Cell a fundamentally different approach: small-molecule chimeras that hijack endoplasmic reticulum (ER)-associated degradation pathway (ERAD) for targeted degradation of transmembrane proteins.
Core concept
Since most TM proteins are folded on the ER membrane during synthesis, the authors hypothesized that engaging ERAD could enable efficient degradation of TM targets. ERAD is initiated by poly-ubiquitination of the target protein by ER-membrane-associated E3 ligases such as SYVN1 (the mammalian homolog of yeast Hrd1), followed by retrotranslocation to the cytosol via the p97/VCP ATPase and subsequent proteasomal degradation. By designing bifunctional chimeras that simultaneously bind SYVN1 and a TM target protein, the authors aimed to redirect ERAD toward selective degradation of therapeutically relevant TM protein PD-L1. These chimeric molecules were termed ERAD-engaging chimeras (ERADECs).
Discovery of desonide as a SYVN1 ligand
The story began with an accidental discovery. While investigating how desonide (an FDA-approved corticosteroid) degrades the mutant huntingtin protein (mHTT) in Huntington’s disease, the authors found that knockdown of SYVN1 completely abolished desonide-induced mHTT depletion. Direct biophysical interactions between desonide and SYVN1 were confirmed by SPR, MST, and ITC, all showing binding at low micromolar Kd. Structure-activity analysis revealed that the 1,3-dioxolane moiety of desonide is critical for SYVN1 binding, as the analog prednisolone, which lacks this feature, did not interact with SYVN1.
Design and potency of PD-L1 ERADECs
For proof of concept, the authors connected desonide to BMS-202, a known PD-L1 ligand, through various linkers. Remarkably, 10 out of 13 P-ERADECs profoundly reduced PD-L1 levels at pM to nM concentrations in MDA-MB-231 and A375 cells. Most effective compounds exhibited sub-nM DC50 and greater than 90% Dmax. Compounds with exclusively flexible PEG linkers (P5 to P7) failed to lower PD-L1, suggesting that a certain degree of rigidity and orientation is required. In humanized NOG mouse xenograft models, intravenous administration at 2.5 or 5 mg/kg significantly inhibited tumor growth, lowered PD-L1 in tumor tissues, and increased T cell infiltration. The effects were abolished in SYVN1 KO tumor xenografts and were not influenced by GR inhibition, confirming the predicted mechanism in vivo.
Mechanistic validation
The mechanism of action was probed in several assays. Knockdown and knockout of SYVN1 abolished PD-L1 depletion, suggesting dependency on SYVN1. The proteasome inhibitor MG132 and the p97/VCP inhibitor CB5083 blocked degradation, while the lysosome inhibitor chloroquine had only a mild effect. Control compounds lacking either the PD-L1 or SYVN1 ligand were inactive. Through AlphaFold2-guided docking and mutagenesis, the authors identified a binding pocket within TM helices 3 to 8 of SYVN1, with Tyr227 forming a key hydrogen bond with desonide. The Y227F mutation abolished both SYVN1 binding and degradation. Ternary complex formation was confirmed by HTRF, coIP, and BiFC, with strong cooperativity (α = 38.3). Proteomics showed excellent selectivity, since PD-L1 was the most significantly lowered protein out of more than 6,000 detected, with no signs of ERAD inhibition or ER stress.
ERADEC vs PROTAC vs inhibitor vs antibody
The P-ERADECs were tested head-to-head against the clinically used PD-L1 antibody atezolizumab and the small-molecule PD-L1 inhibitor BMS-202 in humanized NOG mouse xenograft models. In addition, PD-L1 PROTACs have been published recently, providing opportunity for indirect comparison.
Tumor weights in most P-ERADEC-treated groups were lower than those of mice treated with the PD-L1 antibody atezolizumab or the small-molecule PD-L1 inhibitor BMS-202, suggesting that PD-L1 degradation via ERAD provides a functional advantage over antibody-mediated blockade or small-molecule inhibition. The cellular potency also compares favorably to published PD-L1 PROTACs recruiting cytosolic E3 ligases, which showed DC50 above 1 μM and approximately 60% Dmax. The authors attribute this difference to PD-L1 being folded in the ER, making it more accessible to the ERAD machinery than to cytosolic E3 ligases.
My comments
Interestingly, ERADEC degrader was able to achieve sub-nM DC50 despite only micromolar affinity (by SPR and ITC) of the SYVN1 warhead, plausibly explained by high ternary complex cooperativity (α = 38.3) and efficient (fast kcat) catalytic mechanisms. The onset of degradation is relatively slow (greater than 12 hours), likely because ERADECs target the ER pool rather than the plasma membrane pool directly. Oral bioavailability has not been demonstrated and is likely to be limiting factor, considering the chemical nature of the SYVN1 warhead (desonide). Although GR involvement was carefully excluded, desonide remains a known GR agonist, and developing more selective SYVN1 warheads would be beneficial.
Thus, there is definitely space for further improvement of the steroid-like SYVN1 warhead, by increasing the binary affinity and phys-chem properties. In addition, with approximately 20 different E3 ligases known to mediate ERAD, there is room for expansion.
From my perspective, ERADECs can be seen as a subcategory of PROTACs that hijack ER-associated E3 ligases. Do we really need another special name for each and every “new” modality? But I get it, marketing is important, science including.
Another aspect I would like to point out is that while authors (as well as several other reviews) claim that degradation of TM proteins is limited or challenging, there are several published examples of PROTACs hijacking cytosolic E3 ligases (CRBN) that achieved potent degradation of TM proteins such as kinases MERTK and EGFR. However, it is likely that certain TM targets will be more prone to degradation by ERADECs. Moreover, as the authors suggested, combination of ERADECs with other modalities such as LYTACs could have synergistic effects by simultaneously targeting the complementary pools of TM proteins, located in ER and plasma membrane.
Share your opinion
Which transmembrane protein would you target with ERADECs? Do you think the slow onset of degradation could be a limitation for therapeutic applications?
Leave your comments under my LinkedIn post here.
Full research paper: https://doi.org/10.1016/j.cell.2026.01.018