Bypassing E3 Ligases by 26S-UIDs: Novel Modality for Targeted Protein Degradation
PROTACs and molecular glue degraders are the front runners in the area of targeted protein degradation (TPD), with several clinical candidates. While they demonstrated their usefulness and applicability on a wide range of proteins with diverse function and structure (many of them being disease relevant or hard-to-drug), probing the intricate mechanisms of biological processes and drugging additional (under-explored) biological targets benefits from the ever expanding arsenal of novel (small molecule) modalities, as each of them has its benefits and limitations.
After recognizing the potential of proximity inducing pharmacology, the field is witnessing a fascinating development as novel modalities with distinct mechanisms of action are being published in the (recent) literature. Small molecules are having a big time.
One of the characteristics and limitations of PROTACs and glue degraders is their dependence on E3 ligase machinery, which brings up the following question. What if we could skip the E3 ligase entirely and deliver targets directly to the proteasome?
A new class of degraders: 26S-UIDs
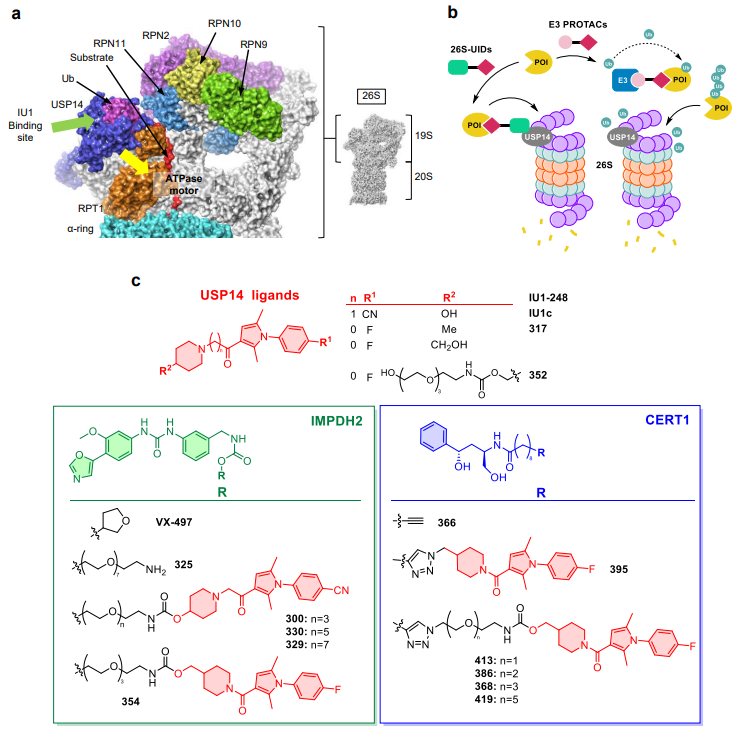
Casasampere, Carneros, Roda, Zuin and colleagues from IQAC-CSIC and IBMB-CSIC in Barcelona now report in Nature Communications a proof-of-concept for small molecule chimeras that direct target proteins to the 26S proteasome via USP14, a proteasome-associated deubiquitinase. They term this modality “26S-UIDs” (26S-targeting ubiquitin-independent degraders).
The rationale for choosing USP14 is compelling. USP14 sits at the base of the 19S regulatory particle, proximal to the ATPase motor that engages and unfolds substrates. USP14 is an allosteric regulator of the proteasome, and its chemical inhibition with the IU1 family of inhibitors enhances 26S activity. Importantly, the initiation of proteasomal degradation (engagement of a flexible fragment by the ATPase engine) is itself ubiquitin-independent. The authors hypothesized that chimeras binding USP14 could localize targets close enough to the ATPase pore to trigger degradation without prior ubiquitination.
Chimera design
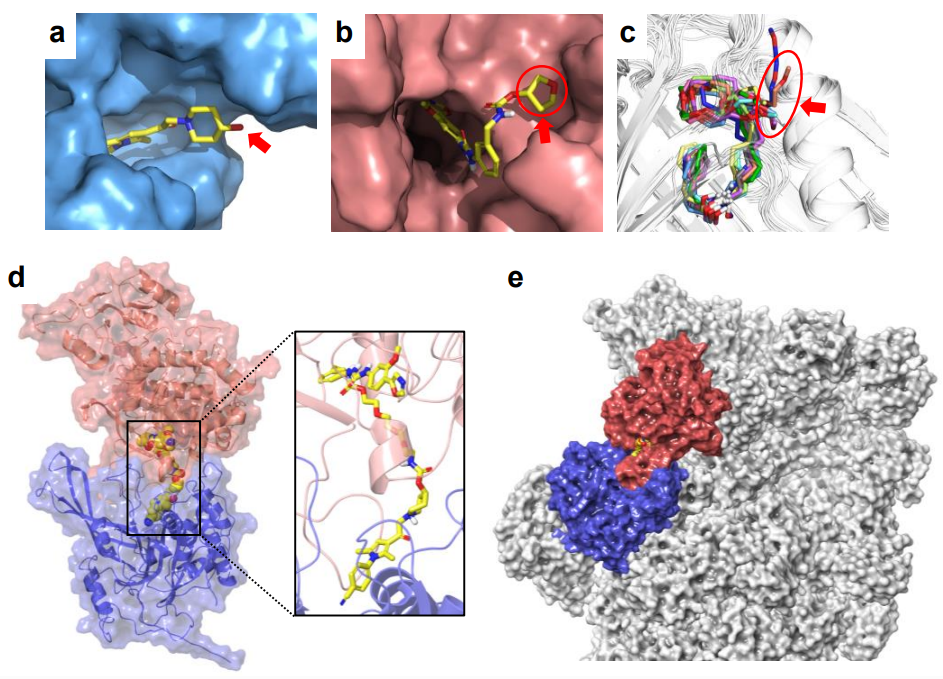
Like PROTACs, each 26S-UID molecule consists of a USP14 ligand, a target protein ligand, and a linker. To recruit USP14, the authors chose inhibitors from the IU1 family. The co-crystal structure of IU1-248 bound to USP14 revealed an exposed hydroxyl group suitable for linker attachment.
For the proof-of-concept, two oncologically relevant proteins were selected for degradation: IMPDH2 (inosine monophosphate dehydrogenase 2), a key enzyme in purine biosynthesis linked to cancer progression; and CERT1, the ceramide transporter shuttling ceramides from the ER to the Golgi, for which no targeted degraders had been reported prior to this work. Structural analysis of each target-ligand complex guided PEG-based linker attachment at solvent-exposed positions, and in silico modeling confirmed that the resulting chimeras could form ternary complexes compatible with the 26S proteasome architecture.
IMPDH2 degradation and the serendipitous discovery of ligand 317
The first series of IMPDH2-targeting 26S-UIDs used VX-497 as the target ligand and IU1-248 as the USP14 binder. Among these, compound 300 induced IMPDH2 degradation at low micromolar concentrations. An interesting twist came when the authors tested compound 317, a derivative of IU1c, as an alternative USP14 ligand. While IU1c had been reported to not inhibit USP14, its binding affinity was never determined. SPR revealed that 317 binds USP14 with comparable affinity to IU1-248 (Kd of 12 and 13 µM respectively) yet barely inhibits its deubiquitinase activity. This turned out to be advantageous: the corresponding bifunctional chimera 354 outperformed the IU1-248-based compounds, achieving complete IMPDH2 depletion at 500-1000 nM in HeLa cells. This suggests that preserving USP14 catalytic function while anchoring the chimera to the proteasome is beneficial for the 26S-UID approach.
However, degradation required 72-96 h, which the authors attribute to a feedback response: the VX-497 moiety triggers upregulation of IMPDH2 mRNA and protein during the initial 24-48 h, counteracting the degradation effect. Importantly, 354 reduced HeLa cell viability with a CC50 of 1.19 µM, while IMPDH2-overexpressing cells were significantly more resistant (CC50 = 34.5 µM), demonstrating that cytotoxicity is driven by target degradation.
CERT1 degradation and ceramide accumulation
Five CERT1-targeting 26S-UIDs using HPA antagonist-derived ligands and compound 317 as USP14 binder were tested in triple-negative (MDA-MB-231) and HER2-positive (MDA-MB-453, BT-474) breast cancer cell lines. CERT1 degradation occurred notably faster than IMPDH2, within 16-24 hours. Since CERT1 transports ceramides from the ER to the Golgi, its degradation led to significantly increased intracellular ceramide levels, particularly for C14-C20 species, consistent with the known substrate preference of CERT1. Compounds 395 and 413 at sub-toxic concentrations sensitized HER2-positive breast cancer cells to lapatinib, hinting at potential therapeutic applications.
Mechanistic validation
The authors provide solid mechanistic evidence. Proteasome inhibitors rescued target degradation (bortezomib and marizomib for IMPDH2; marizomib for CERT1), while the E1 ubiquitin-activating enzyme inhibitor MLN7243 did not prevent degradation of either target, confirming ubiquitin independence. USP14 dependence was demonstrated in Usp14 knockout MEFs, where degradation of both targets was completely abolished. Competition with individual target ligands dose-dependently blocked degradation, confirming productive target engagement. Pulldown assays and computational modeling further validated ternary complex formation compatible with the 26S proteasome structure.
Final comments
26S-UIDs introduce a conceptual shift from E3 ligase recruitment to direct proteasome engagement, which represents an interesting alternative pathway for targeted protein degradation. This can potentially expand the degradable proteome and offer new means for therapeutic interventions.
That said, there are aspects that deserve attention. The long degradation times (72-96 h for IMPDH2 and 24 h for CERT1) are leaving room for potency optimization. I am curious if next generations of 26S-UIDs will be able to compete with the degradation speed of some PROTAC degraders, some of which induce target depletion within a couple of hours.
Share your opinion
Do you think direct proteasome recruitment could become a viable alternative to E3-based degraders? Which targets would you prioritize for this approach?
Leave your comments under my linkedin post here.
Full research paper: https://doi.org/10.1038/s41467-026-71132-5