BRET: State-Of-The-Art Technology for Target Engagement in Living Cells
Direct monitoring of a binding interaction between biomolecules and their ligands is one of the cornerstones of early drug discovery, chemical biology and related disciplines. Assays that allow (semi)-quantitative measurements of the direct binding event, so-called target engagement (TE), became indispensable components of workflows for development or characterization of small molecules, peptides, antibodies, oligonucleotides and other modalities.
There is a wide range of assays for TE evaluation (radioligand displacement, isothermal calorimetry, surface plasmon resonance etc.). However, most of these assays are performed in vitro, using isolated (and often truncated) biomolecules, therefore missing critical physiological factors like membrane permeability, intracellular partitioning, and the influence of biomolecular complexes influencing the ligand occupancy. While these assays are highly valuable, especially for mapping SAR trends. It is highly beneficial to complement them with assays that reflect, as closely as possible, conditions of real physiological environment.
Closer to the physiological conditions are assays that use cell lysate for the detection of TE. However, rupture of the cellular membrane results in a substantial dilution of the cellular milieu, compromising the physiological relevance of these measurements.
The cellular thermal shift assay (CETSA) was the first assay allowing for monitoring of TE in intact cells. Yet, it does not provide quantitative information about the binding affinity and requires elevated temperatures that induce deviation from native physiological conditions.
To address the above mentioned limitations, researchers developed assays based on fluorescence and bioluminescence resonance energy transfer (FRET and BRET). The latter one, BRET, which was developed by researchers at Promega, currently represents state-of-the-art technology for measuring TE in living cells. Over time, the BRET platform developed from “simple” target occupancy quantification to sophisticated mechanism-of-action studies and evaluation of binding kinetics.
How BRET target engagement works
The BRET assay requires two main components: The protein of interest which is fused at C- or N-terminus to a luciferase (typically NanoLuc) and serves as FRET donor (the energy “donor”), and the respective ligand (typically small molecule) which is fused to a fluorescent dye (e.g. BODIPY) and serves as FRET acceptor (the energy “acceptor”). When the luciferase oxidizes its substrate, it enters an excited state which is followed by emission of photons in the visible light spectrum. If the acceptor is sufficiently close (typically <10 nm), the energy of the excited state is transferred via non‑radiative mechanism from the FRET donor to the FRET acceptor. Subsequently, the energy is released from the FRET acceptor via fluorescent emission of photons at distinct (longer) wavelength which can be detected as the assay readout.
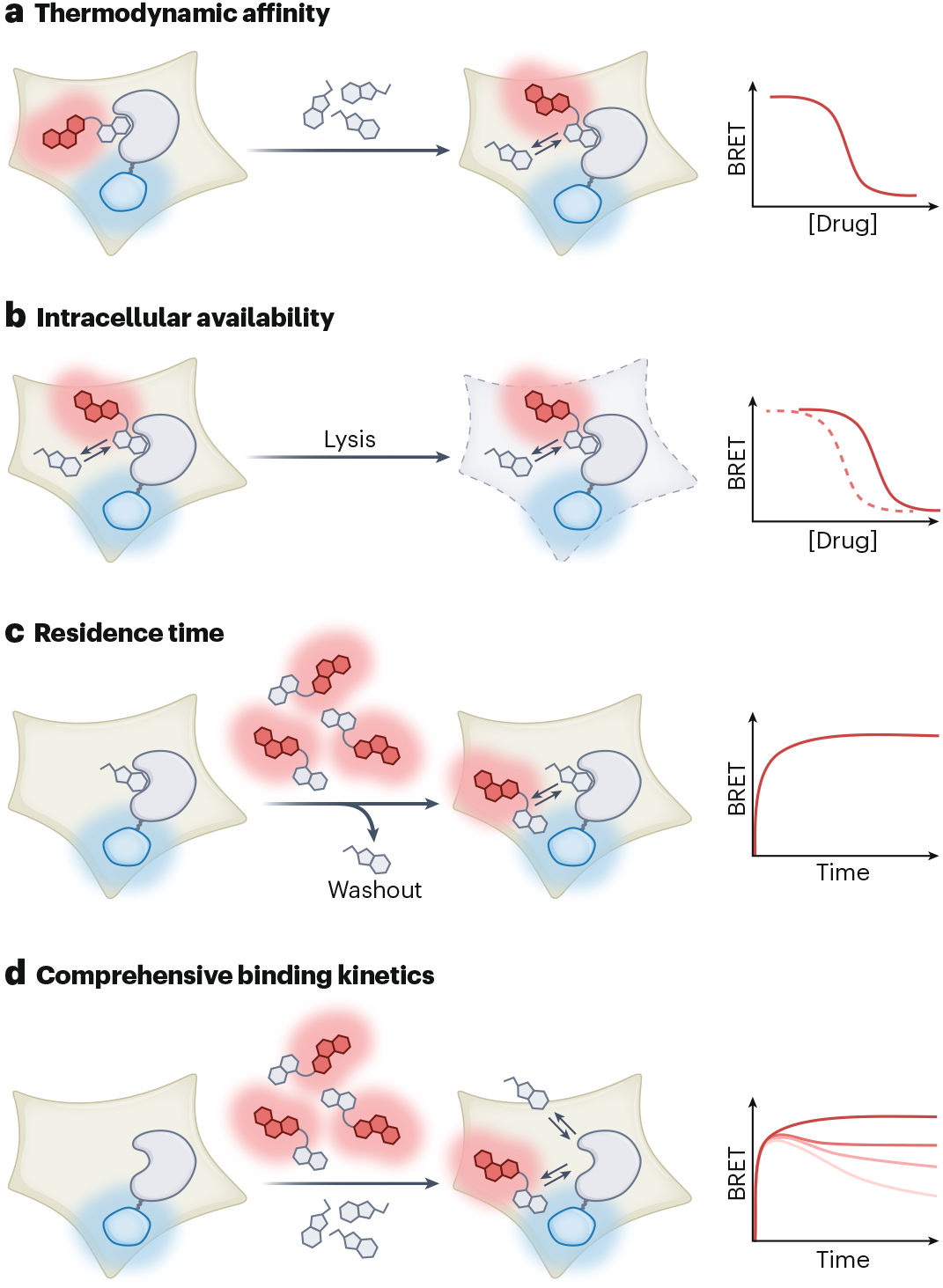
When a ligand binds to the protein of interest and displaces the tracer (ligand fused to the fluorescent dye), the BRET signal decreases. This competitive displacement enables quantitative measurement of the ligand affinity directly in live, intact cells at physiological temperatures. Of note, the BRET assay can detect only binding of ligands that cause tracer displacement.
BRET tracer design
Developing BRET tracers for intracellular TE requires a small-molecule ligand with a sufficient affinity which is decorated with a suitable fluorophore, which is attached in a way that doesn’t interfere with the binding to the protein of interest. To find a suitable attachment for the linker with fluorophore, it is highly advantageous to have information about the binding pose, e.g. from X-ray co-crystal structure, or knowledge of SAR. Alternatively, one can leverage co-folding software such as AlphaFold 3 and Boltz-2.
Finding an optimal linker is essential for achieving a close proximity between the luciferase (FRET donor) and fluorophore (FRET acceptor). In addition, modification of the linker chemistry can be used to adjust physicochemical properties and to ensure sufficient solubility and membrane penetration. Linkers of known potent tracers often employ C3-C6 alkyl chains or 1-3 PEG units, representing a good starting point for development of new tracers.
Residence time and kinetic selectivity
One of the main advantages of BRET assay is the ability to measure binding kinetics in real time, which is particularly relevant for open biological systems where drug concentrations are dynamic. Compound washout experiments in live cells enable quantification of target residence time and kinetic selectivity among related proteins or possible off-targets.
The discordance between thermodynamic and kinetic selectivity can be striking. For CDK inhibitors, compounds with similar equilibrium affinity across the CDK family showed robust durability at CDK6 but rapid dissociation from CDK2 and CDK7. Even more clinically relevant, BRET residence time assays revealed that imatinib resistance in certain BCR-Abl mutants arises through accelerated drug dissociation rather than reduced binding affinity. This finding suggests that kinase inhibitor residence time may underlie drug resistance and could mandate altered dosing regimens in specific patient subpopulations.
A more recent advance applies Motulsky-Mahan analysis to BRET data, enabling simultaneous quantification of association and dissociation rates in cells. This was successfully applied to establish structure-kinetic relationships for a series of bromodomain inhibitors and may serve as a template for extending kinetic TE analysis into other target classes.
Kinome-wide profiling
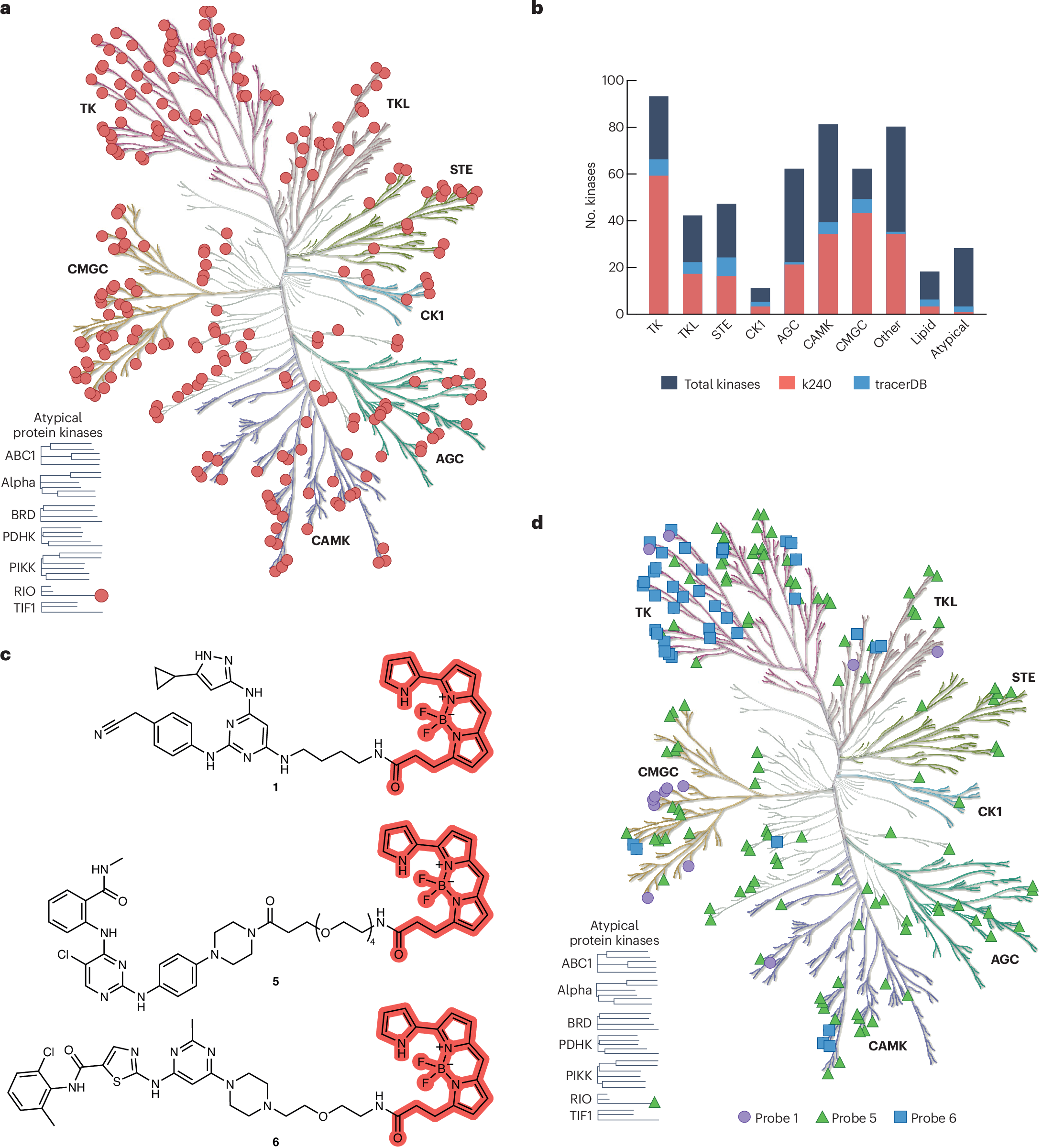
Protein kinases were among the earliest targets adapted by the BRET technology. Leveraging promiscuous kinase inhibitors that target the conserved ATP binding pocket, it was possible to cover 200+ human kinases across most subfamilies using a small set of BRET tracers. This kinome wide profiling revealed a key insight: intracellular ATP can shift kinase inhibitor IC50 by orders of magnitude compared to biochemical assays, underscoring the importance of cellular TE data. The effect of ATP competition can be readily evaluated by comparing engagement profiles in live versus permeabilized cells.
BRET assays have become a standard tool used for development and characterization of (kinase) chemical probes, where on-target engagement and selectivity metrics are key parameters. Recent studies have used the platform for selectivity assessment against panels of up to 240 wild-type kinases. Notably, certain kinases present challenges due to conformational dynamics related to activation states and protein complexes. Modulating intracellular kinase activity using activating mutations has enabled establishment of new assays for additional kinases, as demonstrated for spleen tyrosine kinase and AKT, where gain-of-function variants or phosphorylation status revealed high-affinity inhibitor-bound states.
Quantifying permeability of high-molecular-mass drugs
PROTACs and other beyond-rule-of-5 (bRo5) molecules frequently suffer from compromised cellular permeability, and conventional methods such as PAMPA and CACO-2 often lack the sensitivity needed for reliable measurements. Comparing BRET data from live cells versus lysed or permeabilized cells provides an alternative way for gaining insights about cell membrane penetration.
This approach has been applied in direct-to-biology workflows, where modular assembly of various warhead/linker combinations was systematically built and tested, enabling rapid prioritization of PROTAC permeability and degradation. BRET probes also demonstrated that durable engagement of reversible covalent BTK binders supports increased intracellular accumulation of BTK PROTACs, conferring enhanced target occupancy and degradation. Furthermore, CRBN-targeted BRET probes have been used to establish the role of drug transporter upregulation in PROTAC resistance mechanisms.
In addition, BRET-derived permeability and binding data could serve as a training dataset for machine learning models aimed at predicting behavior of new bRo5 compounds.
Cooperative TE at higher-order biomolecular complexes
Perhaps the most innovative recent applications involve BRET adaptations for monitoring formation of biomolecular complexes, where drug pharmacology is governed by cooperative interactions rather than simple binary binding.
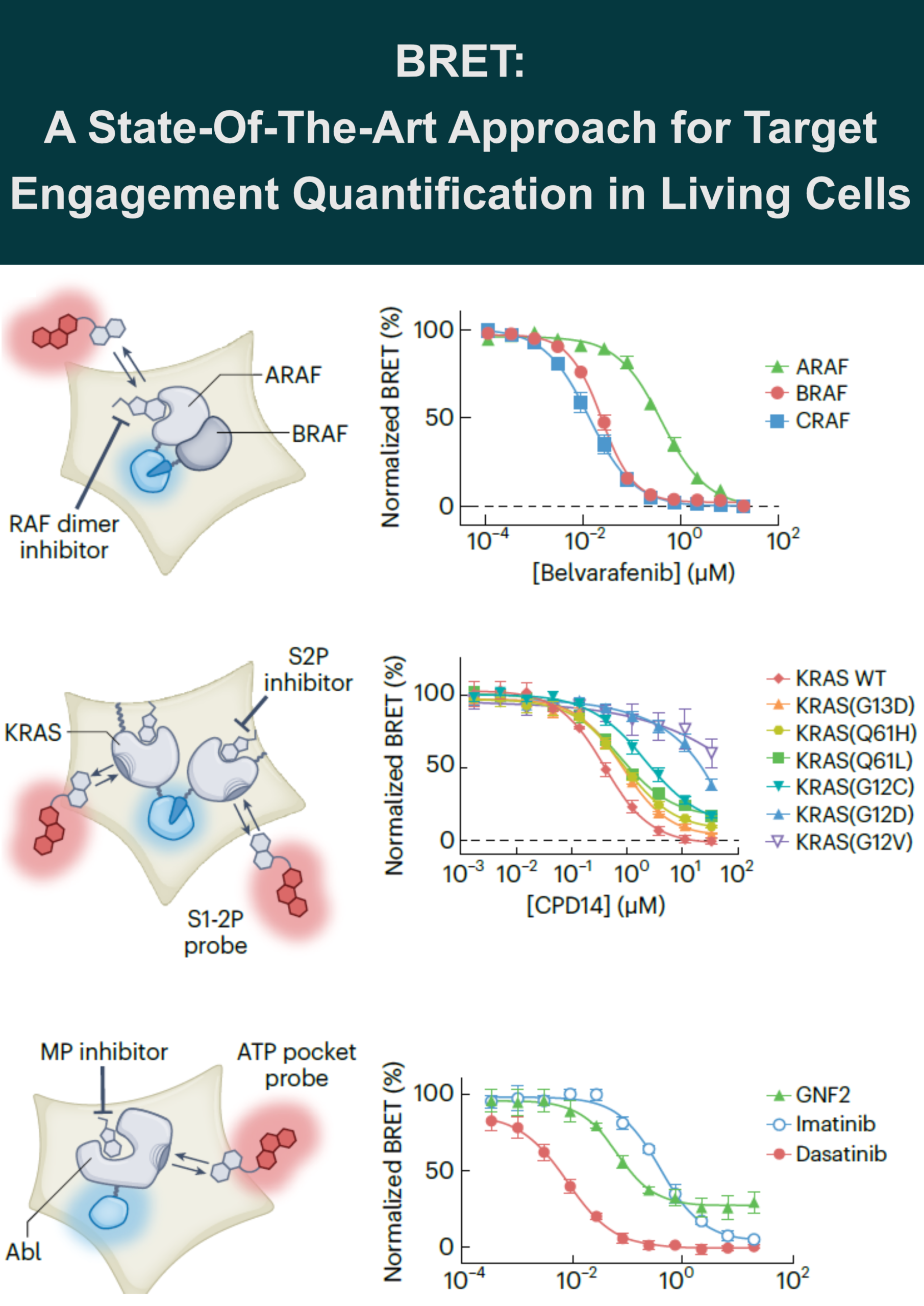
RAF-RAS complexes: To investigate the mode of action for RAF dimer inhibitors and conditionally monitor TE at the RAF–RAS heterotetrameric complex in cells, a novel assay design was established using a combination of enzyme complementation assay (NanoBiT technology) and BRET assay. Each individual protein was tagged with subunits of NanoLuc (SmBiT and LgBiT) to generate a luminescent complex upon RAF-RAS dimerization. This luminescent complex then served as a BRET donor for kinase tracers occupying individual RAF protomers.
This design revealed that clinical-stage RAF dimer inhibitors equally engage BRAF and CRAF protomers but spare ARAF, identifying a potential escape mechanism. The work further demonstrated that RAF dimer inhibitors cooperate with RAS•GTP and stabilize a higher-order RAS-RAF signalosome.
KRAS vulnerability: A BRET probe for the KRAS switch 1-2 pocket (S1-2P) cooperatively engages multimeric KRAS, including common hot-spot mutant alleles. Unexpectedly, this probe revealed that multiple clinical hot-spot KRAS mutations are vulnerable to reversible switch-2 pocket (S2P) engagement using a noncovalent inhibitor (CP14) lacking an electrophilic warhead.
MEK-KSR complexes: A trametinib-derived BRET tracer (Tram-bo) demonstrated that trametinib has longer residence time on the KSR-MEK complex than on free MEK, providing mechanistic insight into its pharmacodynamic effect in vivo. Tram-bo was further applied to profile allosteric engagement across MEK-RAF and MEK-RAF-14-3-3 complexes.
PRMT5-MTA cooperativity: A BRET probe derived from a SAM-cooperative compound was used to quantify TE at the PRMT5-MEP50-MTA-MRTX1719 heterotetrameric complex. Live-cell analysis revealed nearly 100-fold selectivity in MTAP-deleted versus MTAP-intact cells, supporting an uncompetitive mode of engagement and a precision medicine approach at PRMT5.
PROTACs and molecular glues: For event-driven mechanisms, binary target occupancy measurements may not predict degradation efficacy. High-affinity ligands can saturate their target binding sites and suppress ternary complex formation (the “hook effect”). However, evaluation of ternary complex formation and ubiquitination, which may provide stronger correlations to degrader efficacy in cells, can be assessed with protein-protein interaction methods including BRET and NanoBiT (Luciferase enzyme complementation) technology.
Limitations of BRET
The target protein must tolerate a reporter tag such as NanoLuc or NanoBiT, and the method is most suitable for proteins amenable to N- or C-terminal tagging. Importantly, when BRET probes are introduced at or below their apparent Kd for the target, the competitive displacement results provide a close approximation to binding affinity at steady state.
For TE analysis in a pathophysiological setting, the requirement for protein tagging represents a shortcoming. Most BRET studies rely on ectopic overexpression from plasmid-based constructs, which can affect drug-target occupancy measurements. While CRISPR-Cas9 genome editing enables endogenous tagging, many targets may be prohibitively low in abundance. Small NanoLuc subunits (e.g. HiBiT) can facilitate genome editing and provide minimal reporter payload on complex targets such as GPCRs.
The authors envision that detection of endogenous targets via BRET using high-affinity antibodies or nanobodies could ultimately enable quantitative TE studies in diverse cell systems, including patient-derived cells, without the need for protein tags or cell engineering.
Expanding proteome coverage
A major challenge for applying BRET to the understudied “dark” proteome is the requirement for existing high affinity ligands. To address this, the authors highlight several emerging strategies:
DEL-to-BRET workflows: DNA-encoded libraries provide bifunctional starting points where the DNA attachment position can be exploited for linker and BRET dye attachment. This was demonstrated for aurora kinase A, with the resulting probe fortuitously providing assays for several understudied kinases.
NanoBRET-ABPP: Chemoproteomic affinity probes functionalized with photoaffinity and click handles can be rapidly converted into BRET probes after target identification. This combined approach identified a novel photostereoprobe for the mitochondrial serine protease CLPP and enabled a paralog hopping strategy from cyclin E1 to cyclin E2.
TracerDB: A freely accessible, crowdsourced database (tracerDB.org) was created to provide open access to BRET probe validation data, including chemical structures, protocols and quality-assessed verification data. The resource depends on active community contributions.
Are you using BRET probes in your workflows? What targets would benefit most from this approach?
Leave your comment under my linkedIn post here.
This blog article was largely inspired by the following manuscript and complemented by my own experience and opinions: https://doi.org/10.1038/s41589-025-02103-y
#DrugDiscovery #ChemicalBiology