Nickel-Mediated Protein Arylation Inside Living Cells
While transition metals drive powerful bond-forming reactions in synthetic chemistry, their use for covalent modification of biomolecules has been largely underexplored and restricted to isolated proteins in vitro. Aryl-metal reagents for protein S-arylation have so far been dominated by palladium and gold complexes, but these systems require sterically bulky phosphine ligands that promote reductive elimination, resulting in monodentate complexes with potentially limited permeability. Critically, their application for S-arylation inside cells or living systems has not been shown.
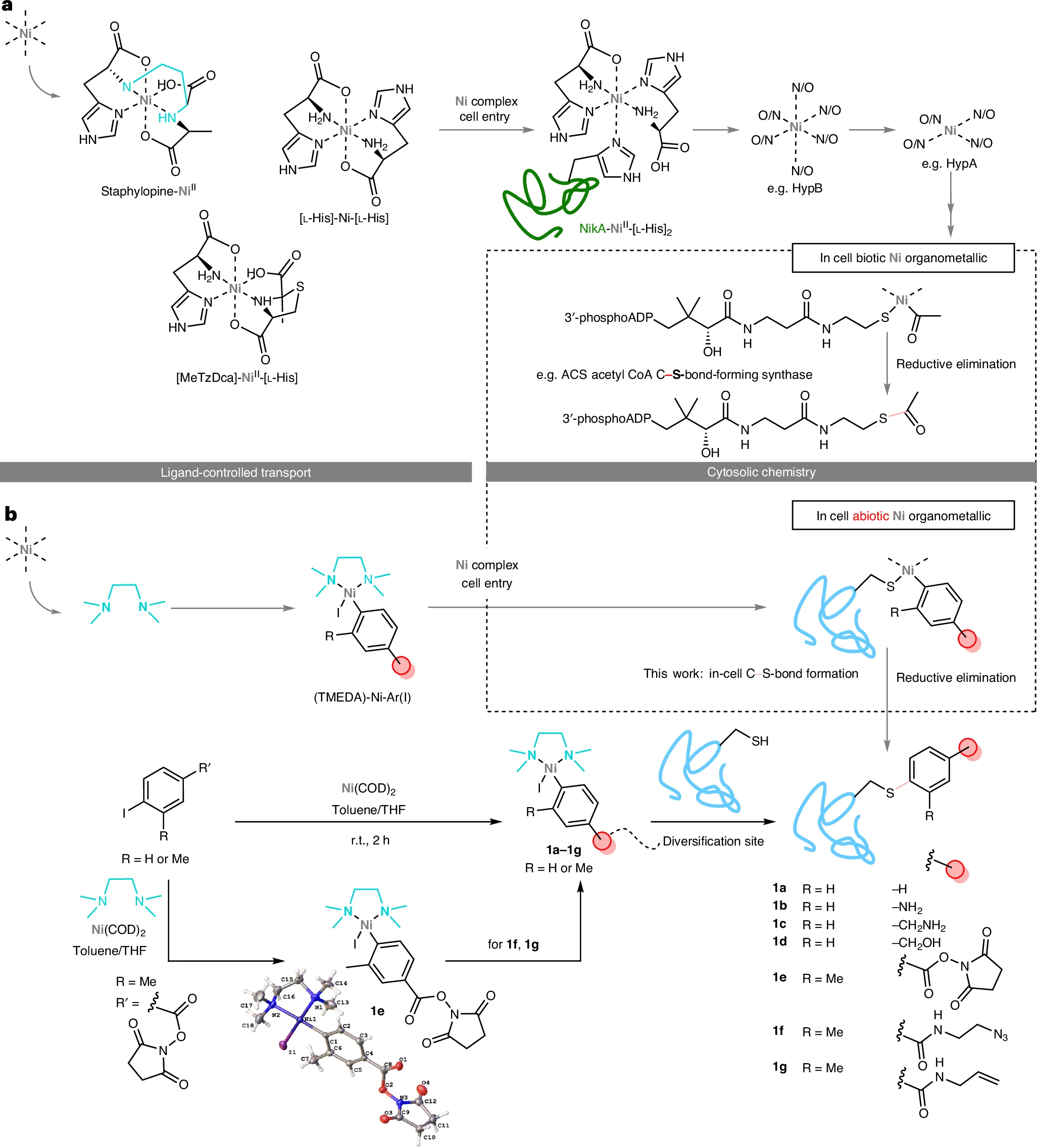
Yet nickel, an essential micronutrient driving diverse enzymatic activities, offers an intriguing alternative. Nickel centres are known in rare biocatalytic systems (such as acetyl-CoA synthase) that mediate reductive elimination to form C-S bonds. In these natural systems, nickel is labile and transportable, with specific helper proteins mediating careful localization through flexible N/O-type ligand coordination that “hands over” the metal while preventing unwanted side reactions.
Xiaping Fu, Weibing Liu and colleagues from the Rosalind Franklin Institute, the University of Oxford and the MRC-University of Glasgow Centre for Virus Research now report in Nature Chemistry a biomimetic strategy inspired by this natural ligand control. By chelating aryl-nickel reagents with biocompatible ligands, they achieve what they call “biocompatible ligand balancing.” maintaining sufficient reactivity for site-selective protein S-arylation while minimizing toxicity, enabling the first demonstration of organometallic C-S bond formation inside living prokaryotic and eukaryotic cells.
Biomimetic design
The core idea of “biocompatible ligand balancing” is to chelate nickel with biocompatible ligands that reduce toxicity and prevent unwanted sequestration in the cellular milieu, while still preserving the chemical reactivity needed for C-S bond formation with protein cysteine residues.
After screening multidentate N-, S- and O-type chelators, TMEDA (N,N,N′,N′-tetramethylethylenediamine) emerged as the optimal ligand. Of note, TMEDA is present in the air-stable Doyle precatalyst and is structurally reminiscent of the alkyl-linked bis-amine found in staphylopine. By mixing TMEDA with Ni(COD)₂ and the corresponding aryl iodide, researchers readily assambled Aryl-nickel reagents (TMEDA)-Ni-Ar(I) (compounds 1a to 1g). Importantly, the NHS-ester variant (1e) allowed chemoselective diversification with amine nucleophiles to install functional groups such as azide (1f) or terminal olefin (1g), without disrupting the nickel complex, prior to subsequent thiol-selective reaction at the metal center.
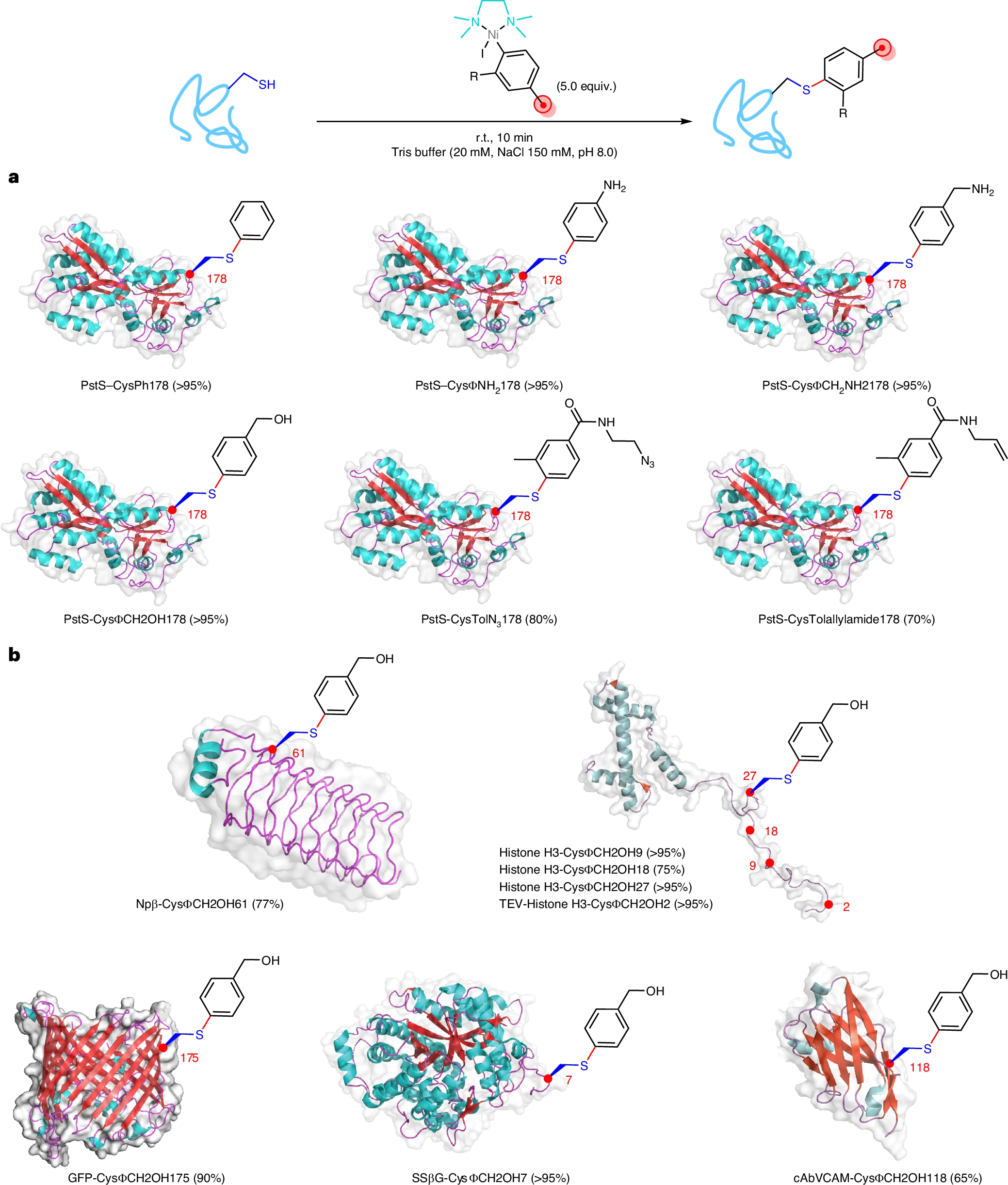
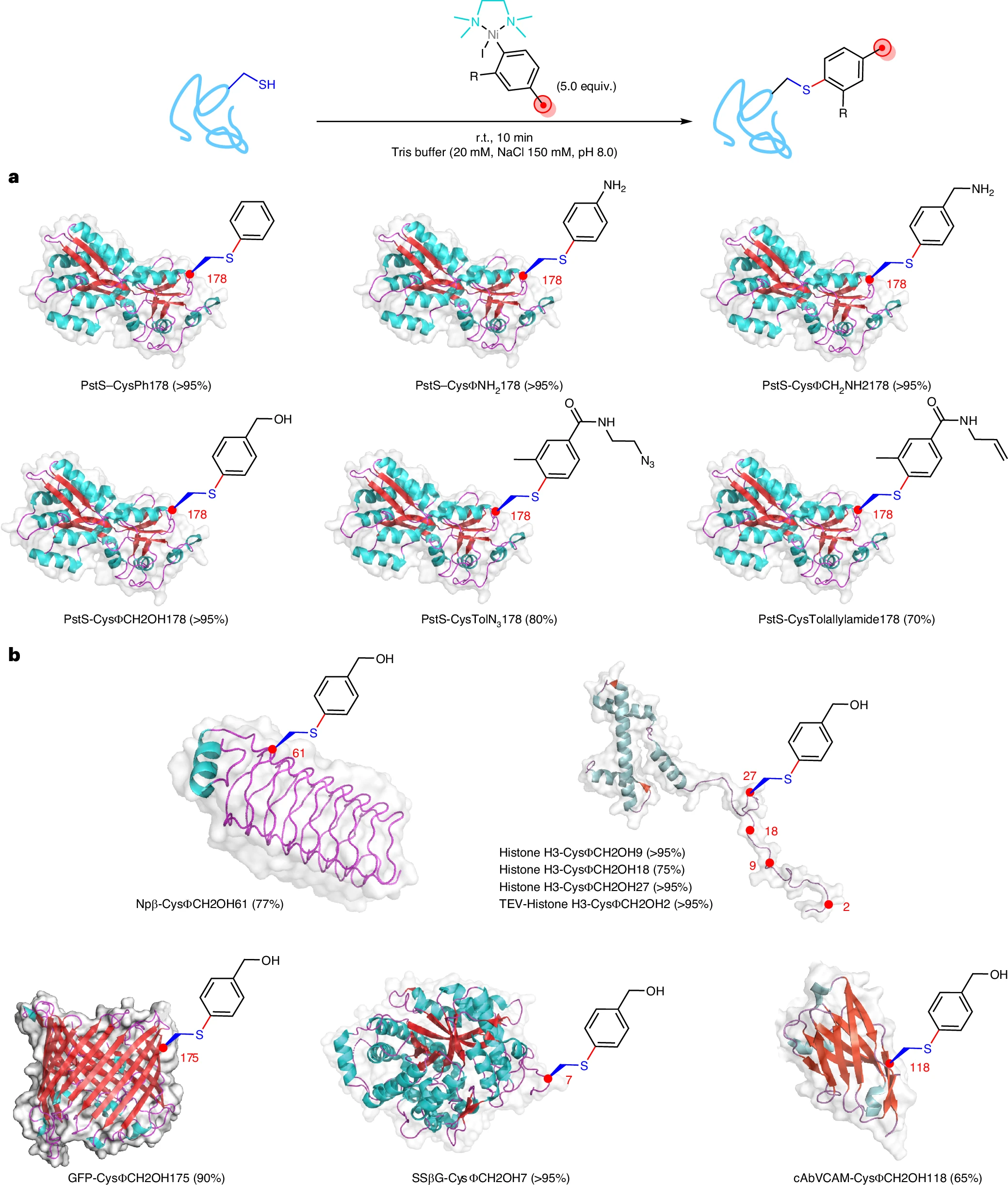
Efficient in vitro protein conjugation across diverse protein folds
Using the model protein PstS, the authors observed rapid and efficient S-arylation at Cys178 with just 5 equivalents of reagent within 10 minutes, achieving conversions above 95% for phenylated variants. Importantly, the system proved broadly applicable across a variety of representative protein folds and functions, including small α-helical histone H3, β-helical pentapeptide repeat protein Npβ, β-barrel GFP, (αβ)₈-barrel glycosidase SSβG, and a nanobody (cabVCAM) containing an internal disulfide. All showed good modification (up to >95%).
Ligand balancing delivers remarkable biocompatibility
The critical test came with cellular toxicity. Non-chelated nickel sources (Ni(OAc)₂, NiSO₄) showed significant toxicity in CHO and HeLa cells at concentrations as low as 0.1 mM. By contrast, the TMEDA-chelated aryl-nickel reagent 1f exhibited negligible cell death even at concentrations up to 2 mM. This dramatic difference confirmed the central hypothesis. Fine-tuning the Nickel coordination is the key determinant of biocompatibility.
Finally, Intracellular protein arylation was then demonstrated in both prokaryotic and eukaryotic cells. In E. coli overexpressing PstS-Cys178, incubation with reagent 1a achieved up to 40% intracellular conversion. In CHO cells treated with 1f, concentration-dependent proteome labelling was confirmed by both western blot analysis and fluorescence confocal microscopy, with transcellular staining observed across cytoplasm and nucleus.
Unprecedented depth in live-cell cysteinome profiling
Another significant result is the depth of cysteinome coverage achieved in living cells. When HEK293T cells were treated with 1f (1 mM, 1.5 h), a dual proteomic workflow combining open search methods and offset-directed search confirmed arylation as essentially the only induced modification, with cysteine selectivity of >90% to >98%. In a single run using nano-LC two-dimensional fractionation, the authors identified 10,982 modified cysteines from 4,963 proteins. This essentially doubles the combined Cys-bearing protein coverage from all previous in-cell profiling datasets accumulated over the past decade (four known datasets covering approximately 13,400 unique cysteines in total). Of the proteins covalently modified by 1f, 83% are classified as “non-ligandable” according to the DrugBank database, and the reactivity showed no strong biases towards active sites, metal-binding sites, or zinc-finger sites.
The dynamic range of detection was also remarkable, spanning more than four orders of magnitude. The method detected not only highly abundant cytoplasmic proteins but also very low-copy-number proteins, and even viral proteins such as SV40 large T antigen and splice variant isoforms of adenovirus E1b that are present at very low abundance.
Tracking intracellular pathogens
To test the limits of sensitivity, the authors applied the method to cells infected with two intracellular pathogens: the obligate intracellular bacterium Chlamydia trachomatis and the alphavirus Sindbis virus (SINV). In C. trachomatis-infected HEK293 cells (at a low MOI of <0.1), more than 500 emerging bacterial proteins were detected, and nine specific cysteine sites from eight bacterial proteins were trapped at 24 hours post-infection. These included OmpA (the major outer membrane protein), ribosomal proteins and, importantly, stage-specific proteins such as DksA and Euo that mark the replicative body (RB) to elementary body (EB) differentiation, a critical and poorly understood transition in the chlamydial developmental cycle.
In SINV-infected cells, the method successfully trapped cysteine sites in two key non-structural proteins: Cys723 in nsP2 (the dual helicase/protease, adjacent to Pro726, a well-characterized non-cytopathic mutation site) and Cys288 in nsP3 (located at the heart of a conserved zinc-binding domain essential for viral replication). The consistent arylation of Cys288 as infection progressed suggested a “privileged” residue and a putative antiviral target. This represents perhaps the first example of transition metal-based direct targeting of a critical pathogen protein in living cells.
My comments
There are some limitations worth noting. The precise arylating species that is functional in cells may arise in situ, and the mechanistic details of cellular uptake and intracellular speciation remain to be fully elucidated. The current work also does not yet address the “undruggable” proteome in a therapeutic sense.
Share your opinion
What applications or modifications do you see for this technology beyond cysteinome profiling? Could ligand-balanced organometallic reagents become tools for covalent drug discovery?
Full article: https://doi.org/10.1038/s41557-025-02017-1