Rewriting Chemistry Textbooks: From Alkene to Alkyne via Hetero-Diels-Alder Reaction
Although Hetero-Diels-Alder will play a central role, I am not going to speak about gender. Not today.
Instead, I would like to highlight another major advancement in organic chemistry that enables synthesis of alkenes from alkynes.
Alkynes serve as versatile building blocks in organic synthesis, drug discovery, and materials science. We chemists use them for click chemistry, cross-coupling reactions, and construction of complex molecular architectures. Yet despite their broad utility, alkynes suffer from markedly lower commercial availability compared to alkenes (30k vs 204k compounds; 6.0M vs 7.9M reported reactions). While (E/Z selective) reduction of an alkyne to an alkene is relatively established chemistry, the reverse transformation has been limiting.
The most established approach dates back to Markovnikov’s 1861 halogenation-elimination protocol, which requires strong bases or high temperatures. These harsh conditions severely limit functional group tolerance and preclude late-stage applications on complex molecules. The fundamental challenge of this synthetic transformation is twofold: the dehydrogenation is thermodynamically disfavored, and the much stronger vinylic C-H bond must be selectively cleaved over the weaker allylic C-H bond.
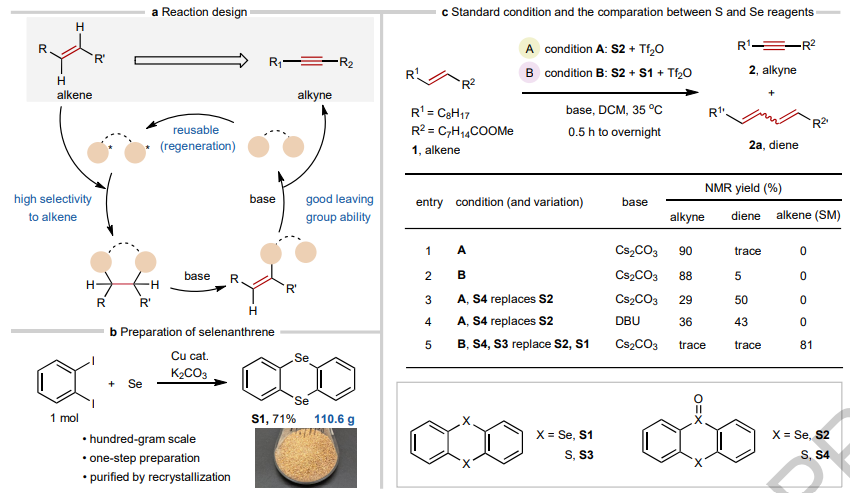
Ning Jiao and colleagues from Peking University now report in Nature a practical (and very creative) method for converting alkenes directly to alkynes under mild conditions, using a recyclable selenanthrene reagent.
Reagent design and reaction development
The authors were initially inspired by the unique dual-heteroatom structure of thianthrene, which can undergo cycloaddition with unsaturated systems and is easily regenerated. The authors therefore designed selenanthrene (S1) by replacing the sulfur atoms in thianthrene with selenium for its high reactivity and ability to form an excellent leaving group. The reagent is readily accessible on hundred-gram scale through a single-step copper-catalyzed reaction followed by recrystallization.
The oxidized form, 5-oxo-selenanthrene (S2), proved to be the key active species precursor. Two complementary sets of conditions were developed. Condition A (S2 + Tf2O) features a simpler setup, ideal for oxidation-tolerant or H-abstraction-sensitive substrates. Condition B (S2 + S1 + Tf2O) is more general and suited for complex, functionalized molecules. In dichloromethane or acetonitrile with mild bases such as Cs2CO3 at 35 °C, E-alkenes were selectively converted to alkynes in 80-90% yield.
Critically, under identical conditions, unmodified thianthrene mainly produced conjugated dienes as side products, highlighting the unique advantage of the selenium-based system.
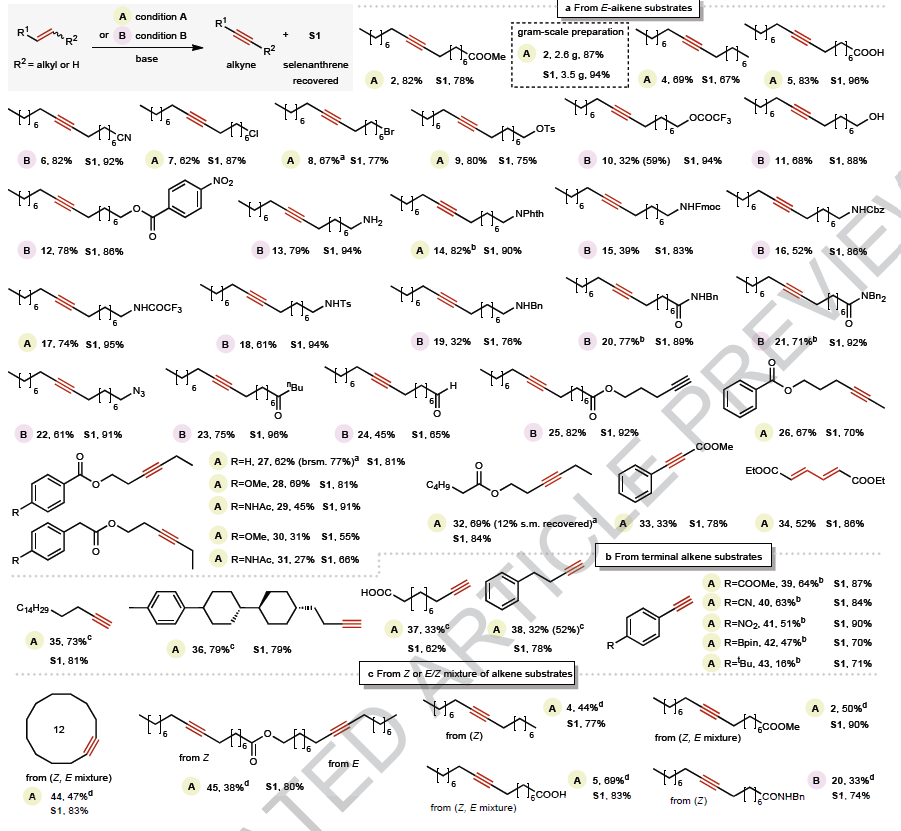
Broad substrate scope and functional group tolerance
Importantly, thanks to the exceptionally mild conditions, the protocol tolerates functional groups that are typically incompatible with conventional methods:
✅ Classical leaving groups: alkyl chloride, bromide, tosylate, Fmoc, trifluoroacetate, epoxide
✅ Functional groups: carboxylic acids, amines, amides, azides, aldehydes, and even pre-existing alkynes
✅ Electron-rich arenes (with reduced efficiency)
The method is suitable for both E-alkenes and Z-alkenes. Terminal alkynes can be achieved by simply changing the base Cs2CO3 for NaOH or KOtBu.
A limitation is represented by substrates with electron-withdrawing substituents which shift the reaction pathway toward conjugated diene formation.
After reaction, the selenanthrene (S1) could be almost entirely recovered and reoxidized with m-CPBA for reuse.
Late-stage applications on complex molecules
Since alkenes are very common functional groups in natural products, the authors didn’t resist the temptation to stress test their new method on complex molecules such as pseudomonic acid D, retapamulin, dasatinib, and a grazoprevir intermediate, as well as cyclic peptides bearing multiple peptide bonds, pyrazines, and other nitrogen-containing motifs. In substrates with sterically differentiated alkenes, the less hindered double bond was selectively converted.
Beyond direct double-bond modification, coupling this approach with alkane dehydrogenation provides a practical route from abundant alkyl groups to valuable alkynes, greatly expanding the range of amenable substrates.
Z/E configuration inversion and sorting
One of the interesting features of this method is its ability to enable interconversion between E- and Z-alkenes, including the thermodynamically uphill E-to-Z conversion, by simply combining this method with stereoselective semi-reduction of alkynes (back) to alkenes.
Remarkably, using a weaker base (NaHCO3), the authors isolated stereospecific alkenyl selenanthrenium salt intermediates that retain the original alkene geometry. Triethylamine selectively promotes elimination from E-alkene-derived intermediates while leaving Z-alkene counterparts untouched. This enables configurational sorting of E/Z mixtures, a long-standing challenge in synthesis due to the minimal chemical differences between E/Z isomers. For example, from a Z/E mixture (1.3:1), the E-isomer was selectively converted to the alkyne (82% yield) while the Z-alkene was regenerated (63% yield) via selective reduction of its selenanthrenium intermediate.
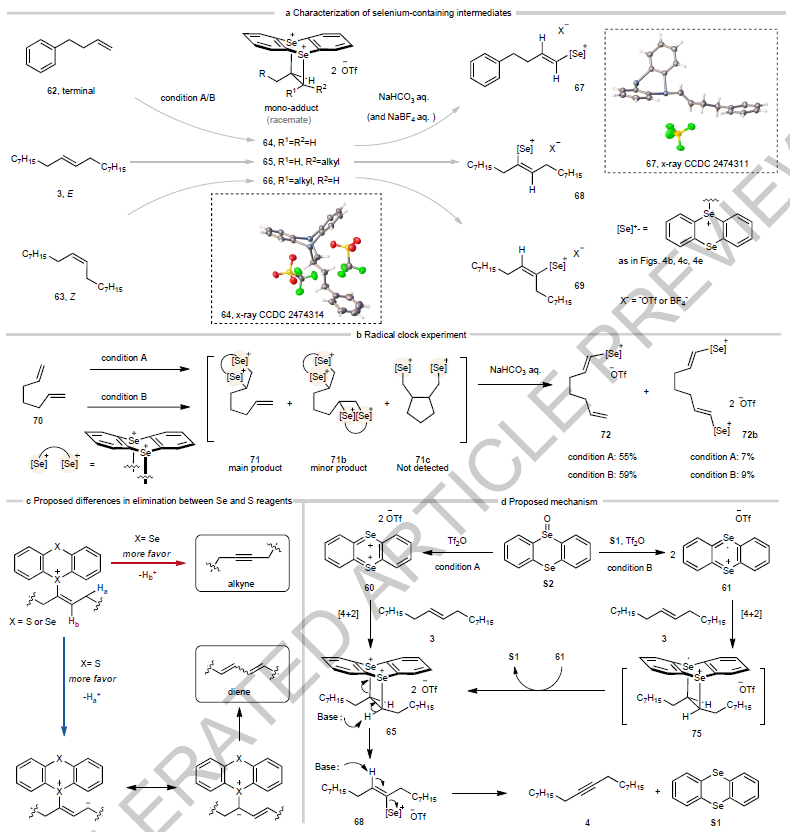
Mechanistic insights
Mechanistic studies revealed two distinct reactive selenium species. A selenanthrene dication is formed under condition A and a persistent radical cation under condition B, both fully characterized by NMR spectroscopy and X-ray crystallography. Despite their different electronic states, both intermediates lead to the same product. A radical clock experiment using 1,6-heptadiene indicates absence of a radical pathway, and supports a mechanism involving hetero-Diels-Alder cycloaddition. (See figure below).
The authors propose that the superior alkyne selectivity of selenium over sulfur reagents stems from differences in carbanion-stabilizing ability. While sulfonium ions more readily stabilize the carbanion formed during allylic proton elimination, favoring diene formation, selenonium ions disfavor this pathway, thus channeling the reaction toward the desired alkyne product.
Conclusion
In my opinion, the authors came up with a unique and very creative way to address this fundamental gap in synthetic chemistry toolbox that has persisted for over a century. Who would guess that alkynes can be generated from alkenes via hetero-Diels-Alder reaction?!
The selenanthrene reagent enables mild and selective alkene-to-alkyne conversion, enabling late-stage functionalization of complex pharmaceuticals and natural products. The recyclability of the reagent, gram-scale applicability, and unique capacity for Z/E sorting and configuration inversion make this a powerful addition to the synthetic methodologies.
🧐 What (complex) molecules would you subject to this new methodology? Are you going to try it out in your chemistry projects?
Leave your comment under my LinkedIn post here.
Full article: https://doi.org/10.1038/s41586-026-10372-3
#SyntheticChemistry #OrganicChemistry