A Unified Platform for Nucleoside Analog Synthesis
Why nucleoside analogs matter and why they’re hard to make
Nucleoside analogs (NAs) are among the most successful drug classes in medicine, with more than 30 currently approved for treating viral infections such as HIV/AIDS and hepatitis, as well as certain cancers. These synthetic mimics of natural nucleosides act as antimetabolites through diverse mechanisms: inhibiting enzymes involved in nucleic acid metabolism, causing chain termination upon incorporation into DNA or RNA, or interfering with epigenetic modification and DNA repair.
Yet despite decades of medicinal chemistry efforts, NA chemical space remains relatively narrow. And one of the main reasons is complex NA synthesis. Traditional synthetic routes begin from chiral pool carbohydrates like D-ribose, require lengthy transformation sequences, and are designed to produce one target at a time. The Vorbrüggen reaction, the standard method for nucleobase attachment, only yields N-linked nucleosides and often gives mixtures of anomers. For context, the initial synthesis of remdesivir proceeded in an overall yield of just 0.6 to 1.5%. Building the large compound libraries that modern high-throughput screening demands has therefore been impractical.
A “nucleobase-last” strategy built on a universal intermediate
Anketell, Britton, and colleagues at Simon Fraser University, in collaboration with Merck, now report a synthetic approach that fundamentally redesigns NA synthesis. The core idea is elegant: construct a single versatile intermediate from cheap, achiral starting materials, then attach the nucleobase in the final step using photoredox chemistry. Because diversification happens last, the entire upstream synthesis needs to be done only once per sugar scaffold, making it ideal for library generation.
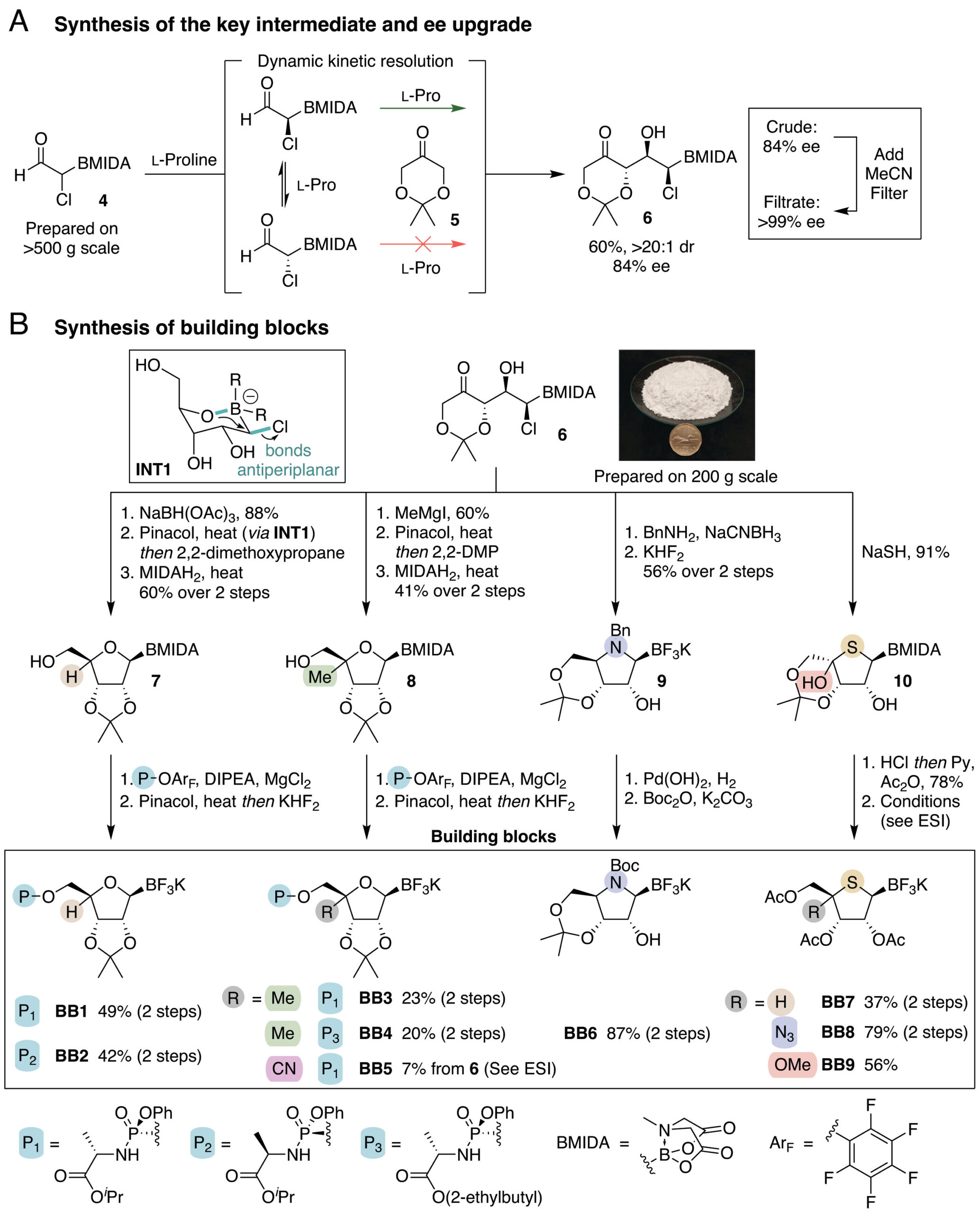
The synthetic route starts with an asymmetric proline-catalyzed aldol reaction between an alpha-chloro boroaldehyde and dioxanone, proceeding through a dynamic kinetic resolution with excellent diastereoselectivity. A serendipitous finding boosted the process further. The racemic form of the product turned out to be nearly insoluble in acetonitrile, enabling a simple filtration step that delivered greater than 99% ee. The authors validated scalability by preparing over 500 g of the aldehyde precursor and 200 g of the enantiopure aldol adduct with minimal modification to the gram-scale protocol.
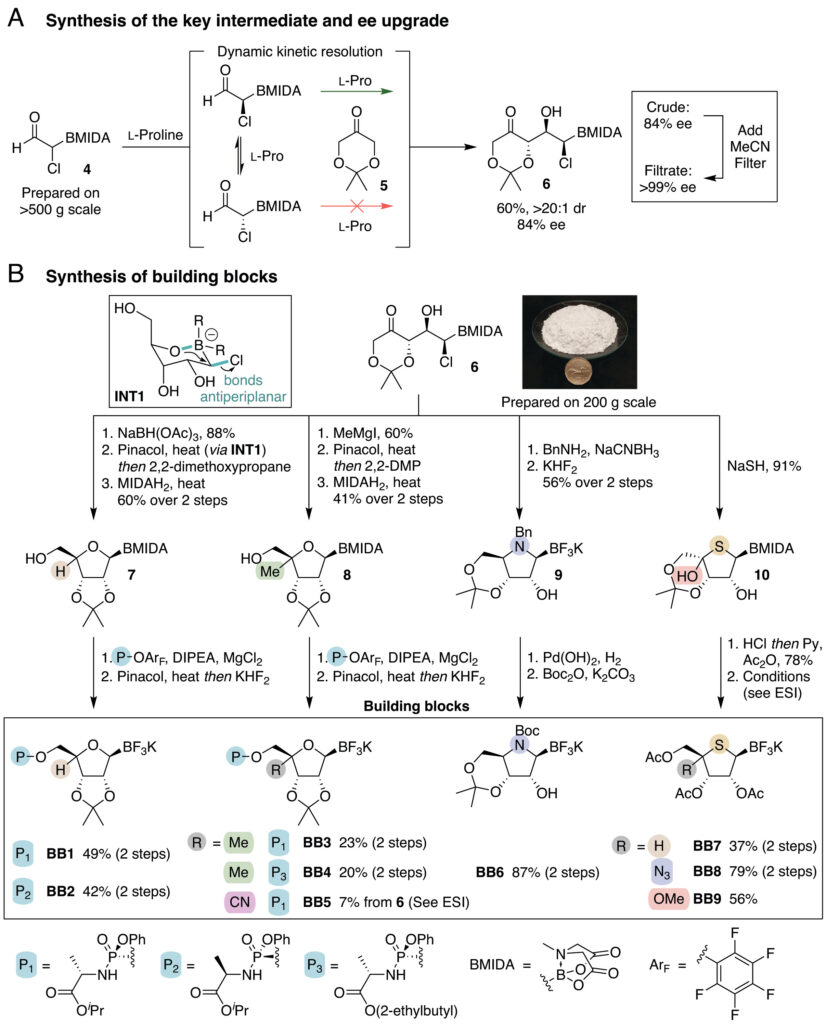
One intermediate, nine building blocks, all major NA classes
From this single key intermediate, the authors branched out into nine building blocks (BB1–BB9) covering all major NA classes. Standard ribose-derived building blocks were obtained through a selective 1,3-syn reduction and an intramolecular Matteson-type cyclization that forms the ribose ring while preserving the boron handle. These were then equipped with ProTide moieties, the phosphoramidate prodrug groups found in approved drugs like sofosbuvir and remdesivir, and a trifluoroborate at the anomeric position, ready for the final coupling.
C4′-modified building blocks with methyl or cyano substituents were accessed via a stereoselective Grignard addition. Iminonucleoside scaffolds were obtained in a single step through reductive amination that triggered spontaneous cyclization. Thionucleoside cores were built by treating the aldol adduct with NaSH, directly furnishing the thiosugar in excellent yield. This remarkable convergence, all from one starting intermediate, is a key strength of the platform.
Photoredox coupling: the diversification engine
The final nucleobase installation relies on photoredox-mediated coupling of the trifluoroborate building blocks. For C-linked nucleosides, an iridium/nickel dual catalytic system proved optimal, while N-linked nucleosides required an acridinium photocatalyst/copper system optimized through high-throughput experimentation. Both methods delivered excellent anomeric diastereoselectivity (greater than 10:1), attributed to steric shielding by the acetonide protecting group. Importantly, both tolerated the complex ProTide functionality.
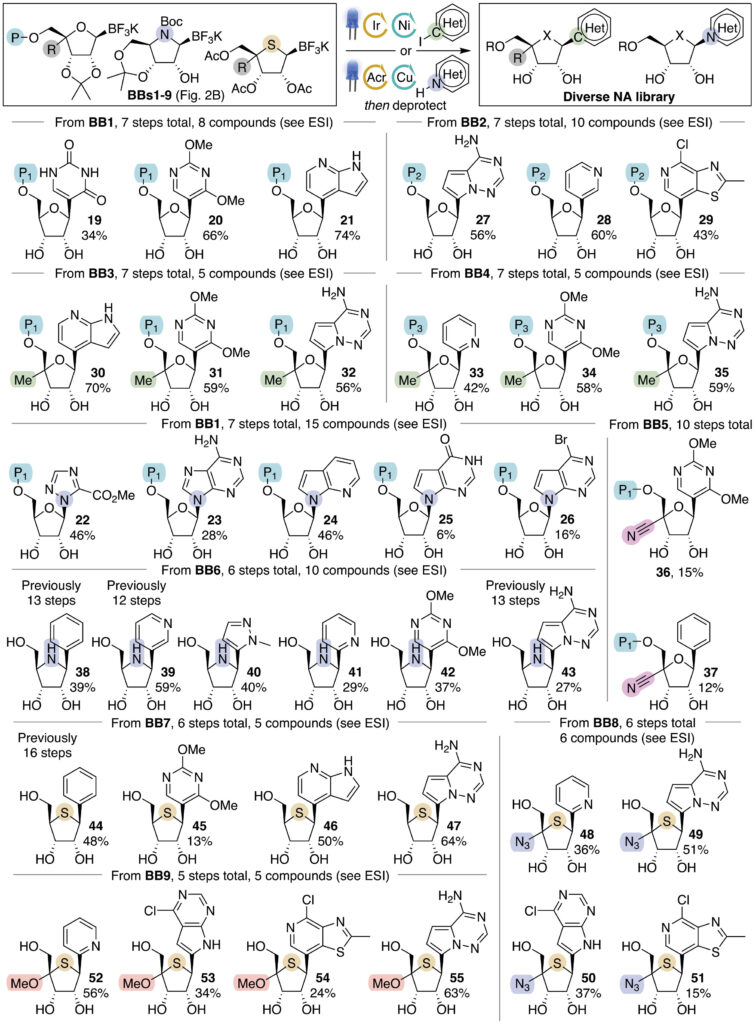
A 71-member library and anti-HIV hits
Using this platform, the team assembled a library of 71 nucleoside analogs spanning ProTides, C4′-functionalized NAs, iminonucleosides, and thionucleosides. All 45 ProTide derivatives are novel. The efficiency gains are remarkable: C4′-modified ProTides were prepared in just 7 steps from the chloroaldehyde. A nanomolar-potency nucleoside hydrolase inhibitor previously made in 13 steps from D-ribose was obtained in 6 steps. A known antiviral iminonucleoside active against influenza was similarly shortened from 13 to 6 steps, and a thionucleoside previously requiring 16 steps was accessed far more rapidly.
Cheminformatic analysis using PCA and k-means clustering confirmed that the library spans three of four identified clusters in known NA chemical space, overlapping well with approved drugs. Screening against HIV-1 in a cell-based replicon assay identified three hits: a C-linked ProTide bearing the remdesivir nucleobase (IC50 = 0.93 µM), an iminonucleoside (IC50 = 1.2 µM), and an N-linked ProTide (IC50 = 0.46 µM), representing screening hits with excellent potency and potential for further elaboration.
Why this matters
This work addresses a longstanding bottleneck in nucleoside drug discovery. By unifying the synthesis of all major NA scaffolds from a general intermediate and enabling late-stage diversification through photoredox coupling, this synthetic approach makes it practical to build the kind of diverse NA libraries that high-throughput screening campaigns require. Moreover, this approach is not only general and divergent, but also highly efficient as it allowed synthesis of complex NA in fewer steps and with better overall yield. Given that nucleoside analogs have already proven their therapeutic value in both antiviral and oncology settings, opening up the vast unexplored regions of their chemical space could accelerate the discovery of next-generation NA-based therapeutics.
Full article: https://www.science.org/doi/10.1126/science.aed6880
Leave your comment under my LinkedIn post here.