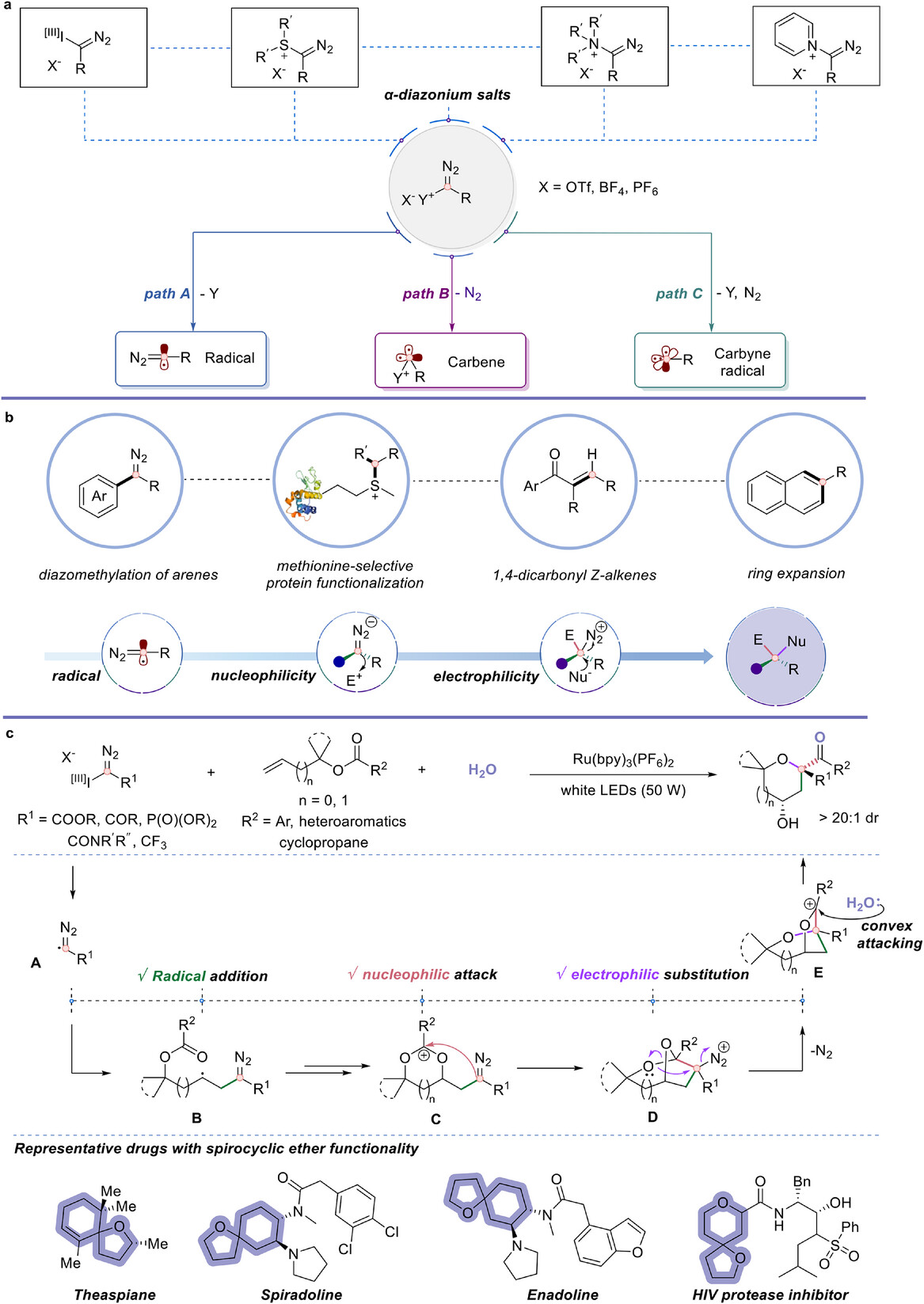
Accessing Spirocyclic Ethers via Chemical Cascade with Carbyne Equivalents
We chemists love building molecular complexity efficiently and in as few steps as possible. This allows us to transform readily accessible compounds into compounds of high value and to explore and access the vast chemical space which is highly relevant to drug discovery, material science and other fields related to synthetic chemistry.
One of the popular approaches for achieving that is the utilization of reaction cascades sequentially generating multiple bonds in one reaction vessel. But how would you build such a chemical domino sequence to form three new bonds around a single carbon?
The answer has two words: Carbyne equivalents!
Carbyne equivalents represent trivalent carbon synthons with three nonbonded electrons, theoretically capable of forming three C-C or C-X bonds simultaneously. Since Fischer’s first carbyne complex in 1973, groups of Suero, Gaunt, Wang, Glorius, Ooi and others have significantly expanded the synthetic utility of these intermediates, enabling C-H functionalization, skeletal editing, protein functionalization, and heterocycle synthesis. Yet, fully exploiting all three reactive modes in a single catalytic reaction cascade forming three σ-bonds has remained scarce.
Jianke Su, Chu Wang, Jie Wu and colleagues from the National University of Singapore and the University of Science and Technology of China now report in Angewandte Chemie a photoinduced cascade that exploits the reactivity of this unique reagent class.
By careful tuning of the radical, nucleophilic, and electrophilic reactivity of carbyne equivalents, the authors developed a methodology for selective construction of densely functionalized spirocyclic ethers from allylic and homoallylic benzoates.
How it works
The cascade begins with photoinduced single-electron transfer (SET) from the excited Ru(bpy)₃(PF₆)₂ catalyst, which reduces the hypervalent iodonium diazo compound to generate an α-diazo radical which undergoes radical addition onto the alkene of the allylic benzoate (radical reactivity, first σ-bond).
The resulting radical intermediate is oxidized to a carbocation, triggering intramolecular nucleophilic attack by the vicinal carbonyl oxygen. This leads to a cyclic intermediate where the carbyne carbon center then acts as a nucleophile (nucleophilic reactivity, second σ-bond).
In the final stage, neighboring group-assisted C-O bond cleavage occurs with an oxygen [1,2]-shift and N₂ extrusion. Here the carbyne fragment functions as an electrophile (third σ-bond), generating a carbocation that is captured by water through diastereoselective ring-opening to give the spirocyclic ether product.
Of note, the proposed mechanism is supported by a thorough mechanistic investigation.
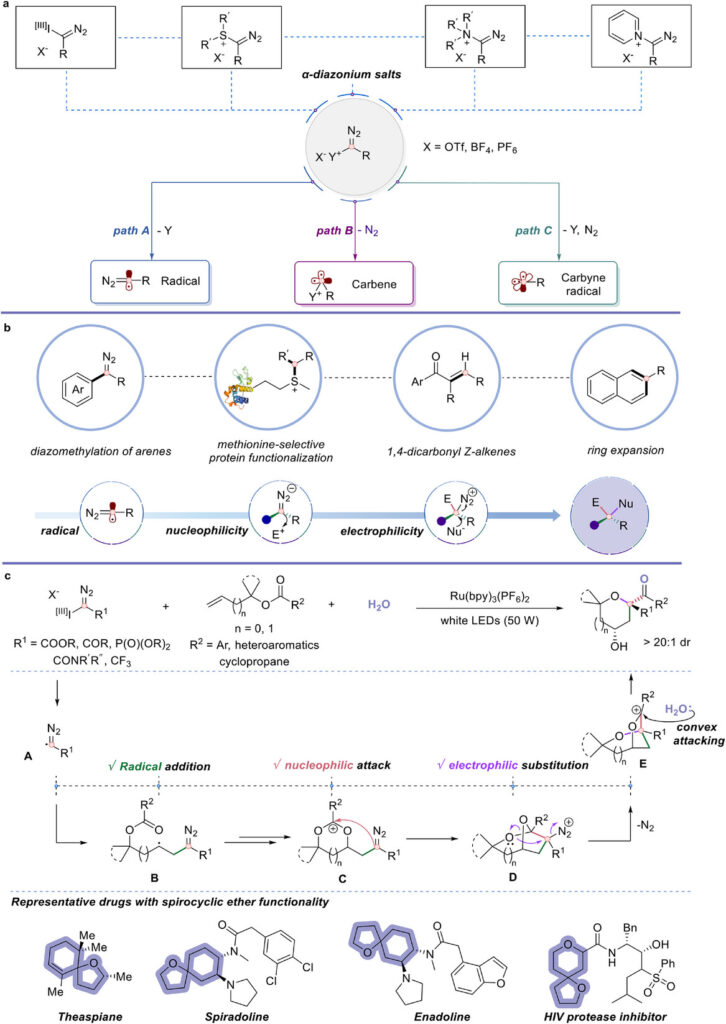
Reaction conditions
The optimized protocol uses carbyne equivalent (see the Figure 2 below), Ru(bpy)₃(PF₆)₂ (1 mol%) as the photocatalyst, NaHCO₃ (2 equiv.), H₂O (2 equiv.) in CH₃CN and 50 W white LED at 0 °C under argon.
The hypervalent iodonium diazo compound 2a performed best among several carbyne precursors.
A practical note: the cheaper Ru(bpy)₃Cl₂ salt delivered comparable yield, and that all tested carbyne precursors (including sulfonium triflates) are bench-stable and readily accessible.
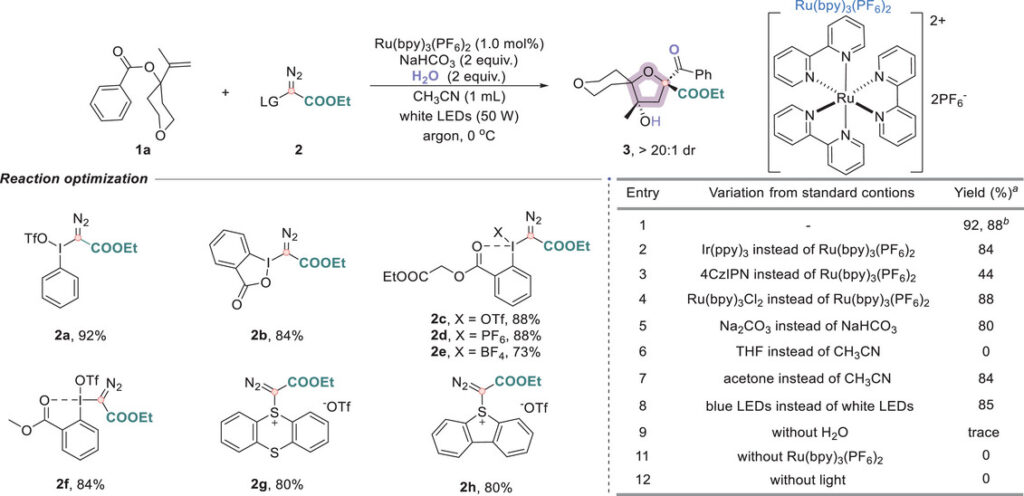
Substrate scope
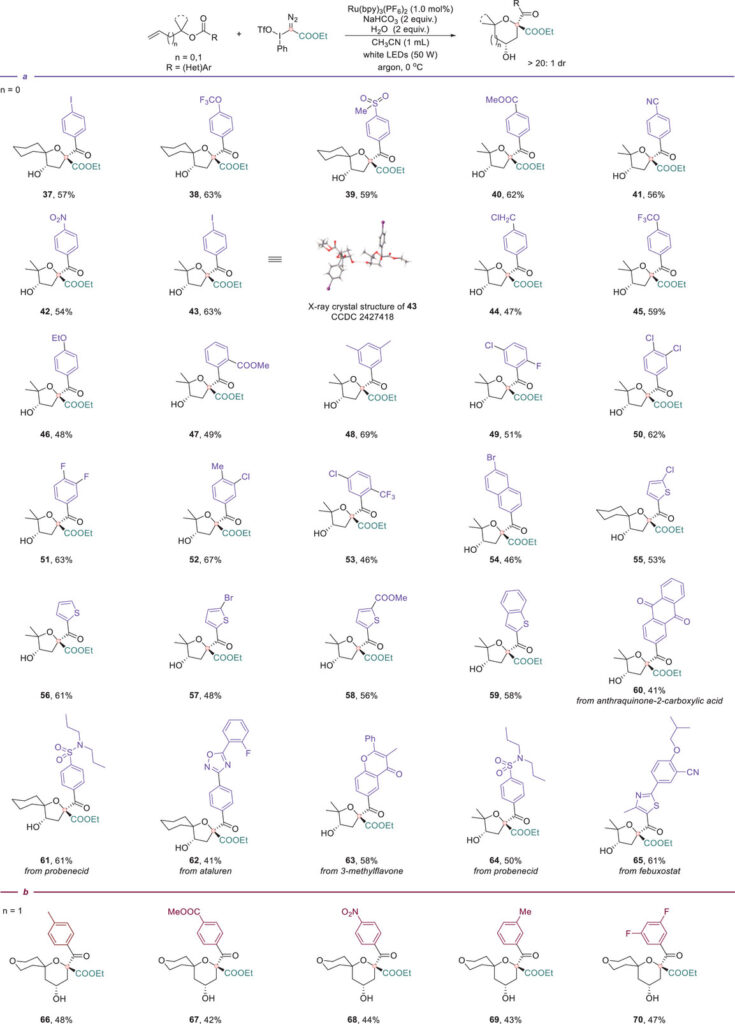
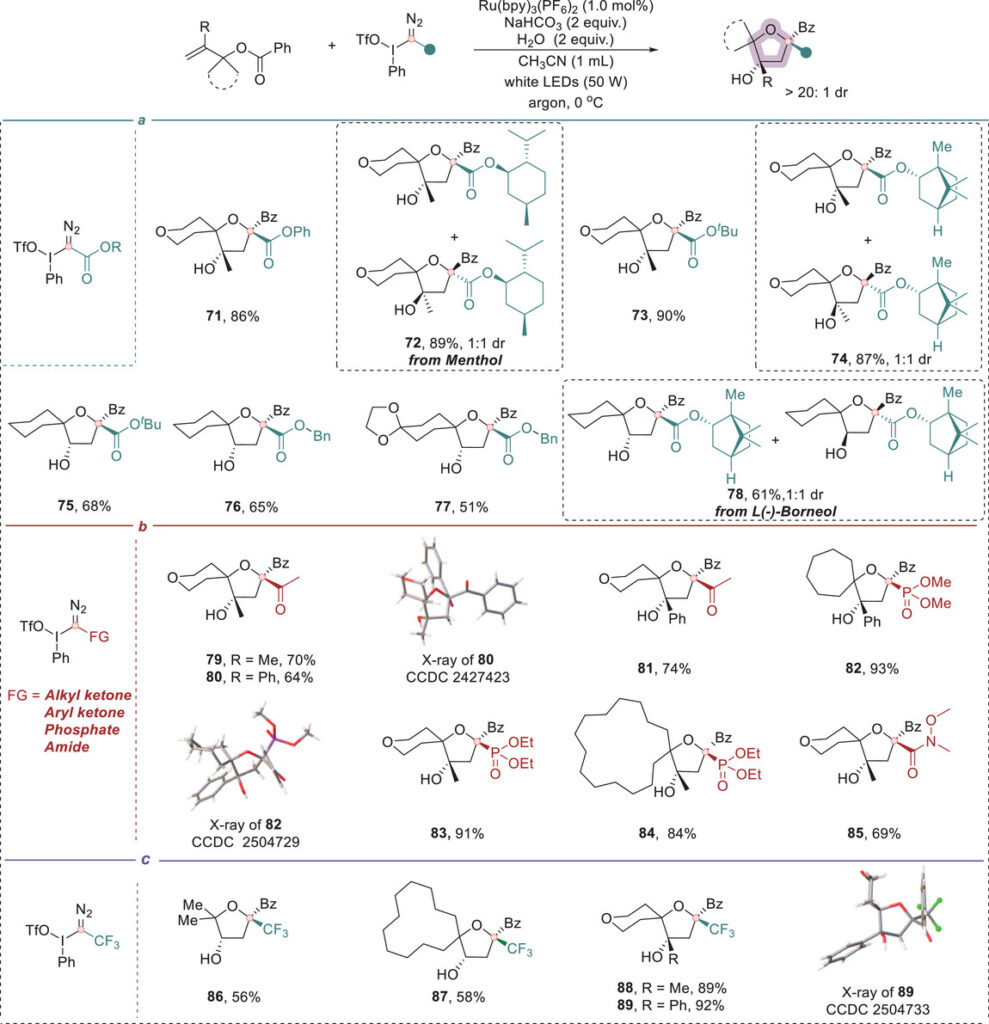
Carbocyclic substrates containing four- to fifteen-membered rings were transformed into the corresponding spirocyclic ethers. Heteroatom-containing rings (O, N, S) delivered dioxaspiro, azaspiro, and thiaspiro scaffolds. A variety of substituents on the ring moiety (methyl, gem-difluoro, phenyl, cyclic ketals) were well tolerated.
The aryl group on the benzoate tolerates substituents of various electronic nature, including electron-withdrawing (-NO₂, -CN, -I, -OCF₃), electron-donating (-OEt), as well as sterically hindered, and heterocycles.
Biorelevant molecules such as probenecid, ataluren, febuxostat, and 3-methylflavone were incorporated into the spirocyclic framework. Natural product-derived substrates (cyrene, corodane) and late-stage functionalization of adamantanone, norcamphor, and menthone further demonstrate the method’s generality. Homoallylic substrates gave tetrahydropyran-fused spirocycles, and trifluoromethylated carbyne equivalents afforded CF₃-containing spirocyclic ethers.
The reaction ran smoothly also at 50 mmol scale in a circulation-flow reactor and provided 9.52 grams of product with 65% yield.
Final comments
The strict requirement for gem-disubstitution at the allylic position (Thorpe-Ingold effect) narrows the substrate scope, and the yield for strained systems such as the oxetane-fused spirocyclic product (37%) leaves room for improvement. It would also be beneficial to have an enantioselective variant of this reaction.
Spirocyclic ether motifs are highly relevant for small molecule drug discovery and new ways to access them efficiently are needed. Besides providing access to this structural motifs, this cascade approach also demonstrates the unique synthetic utility of carbyne equivalents which will likely inspire additional ways for their synthetic application.
Share your opinion
Where do you see additional (undiscovered) application of carbyne equivalents? Are you going to try this protocol in your lab? Let me know how it works!
Full research article: https://doi.org/10.1002/anie.1489528